Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000546.3 | Alternative | 2640 nt | 252–1433 |
| NM_000546.5 | RefSeq Select | 2591 nt | 203–1384 |
| NM_000546.6 | MANE Select | 2512 nt | 143–1324 |
| NM_000546.4 | Alternative | 2586 nt | 198–1379 |
| NM_000546.2 | Alternative | 2629 nt | 252–1433 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenVariant summary: TP53 c.845G>A (p.Arg282Gln) results in a conservative amino acid change located in the DNA-binding domain (IPR011615) of the encoded protein sequence. Four of five in-silico tools predict a damaging effect of the variant on protein function. The variant allele was found at a frequency of 4e-06 in 251444 control chromosomes (gnomAD). c.845G>A has been reported in the literature in individuals affected with a variety of cancers but not fulfilling the classic criteria of LFS or even the LFI (Li-Fraunemi Incomplete) criteria. In our review of the associated literature, the penetrance of Li-Fraumeni Syndrome (0.67) due to this variant appears to be lower than expected (0.8), therefore no conclusions can be drawn from these data. Specifically, this variant was observed in a patient with neuroblastoma and was reportedly inherited from one of her parents, but the family history (a female patient, diagnosed at age 1 with neuroblastoma, and a maternal aunt with lymphoma diagnosed at 44) was not suggestive of Li-Fraumeni syndrome (Chompret 2000). The variant has been also reported as germline variant in several other cancer patients, including lung-, breast-, prostate and colorectal cancer, but without strong evidence for causality (Meric-Bernstam_2016, Tung_2016, Monti_2007, Giri_2019, Stoltze_2018). In addition, in one of these reports a co-occurrence with another likely pathogenic variant has been reported (PALB2 exon 13 deletion; Giri_2019), providing supporting evidence for limited causality. Additional studies are needed to address the penetrance and cancer risks associated with TP53 pathogenic variation in patients outside LFS spectrum. Several publications reported experimental evidence evaluating an impact on protein function, and multiple yeast assays demonstrated that the variant decreased, but did not abolish the transactivation capacity of TP53 and has been reported as a partially deficient (PD) allele (e.g. Monti_2011, Andreotti_2011 , Resnick_2003, Shi_2002). On the other hand, further studies performed in yeast and in human cells, revealed that the variant could also result in a gain of function activity, by interfering with the function of other p53 family members and increasing the expression of genes involved in cell proliferation- and tumor formation (Monti_2003, Shi_2002, Cordani_2011). These data however do not allow unequivocal conclusions about the variant significance. Six other clinical diagnostic laboratories have submitted conflicting clinical-significance assements for this variant to ClinVar after 2014 (i.e. 4 calling it a VUS, while 2 classifying it as likely pathogenic). At-least two of these submissions reflect a re-evaluation from their original assessment in the pathogenic spectrum. Based on the overall evidence outlined above, the variant was re-classified from its initial assessment as Likely Pathogenic at our laboratory and has since retained its classification as a VUS-possibly pathogenic.
The following ACMG criteria was used: PM2_SUP; PM1; PP3; BS3
This missense variant replaces arginine with glutamine at codon 282 in the DNA binding domain of the TP53 protein. Computational prediction suggests that this variant may have deleterious impact on protein structure and function. Functional studies have shown no or partially disruptive effect of this variant on transactivation activity in yeast-based assays (PMID: 11429705, 11896595, 11920959, 12826609, 12909720, 12917626, 21343334) and no effect on the anti-proliferative function of TP53 protein in mammalian cell-based assays (PMID: 29979965, 30224644). This variant has been reported in an infant affected with neuroblastoma with history of lymphoma in her maternal aunt (PMID 10864200). This variant has been observed in an individual affected with lung cancer in his seventies, with family history of breast and prostate cancer in his siblings (PMID: 26787237). This variant has also been observed in individuals affected with breast cancer (PMID: 26976419) and glioneuronal tumor (PMID: 29058119), as well as in an individual affected with colorectal cancer with early-onset breast cancer history in her mother and maternal grandmother (PMID: 29324801). This variant has been identified in 1/251444 chromosomes in the general population by the Genome Aggregation Database (gnomAD). Different variants affecting the same amino acid position (p.Arg282Trp, p.Arg282Pro) are considered to be disease-causing (ClinVar variation ID: 12364, 376659), suggesting that arginine at this position is important for protein function. However, the available clinical and functional evidence is insufficient to determine the role of the p.Arg282Gln variant in disease conclusively. Therefore, this variant is classified as a Variant of Uncertain Significance.
The p.R282Q variant (also known as c.845G>A), located in coding exon 7 of the TP53 gene, results from a G to A substitution at nucleotide position 845. The arginine at codon 282 is replaced by glutamine, an amino acid with highly similar properties. This alteration has been reported in a family in which the proband was diagnosed with neuroblastoma at 1 year of age and the proband's maternal aunt was diagnosed with lymphoma at 44 years of age (Chompret A et al. Br. J. Cancer. 2000 Jun;82(12):1932-1937). Functional studies conducted in yeast have demonstrated partially reduced transactivation activity compared to wild type (Campomenosi P et al. Oncogene. 2001 Jun;20:3573-9; Shi XB et al. Prostate. 2002 Apr;51:59-72; IARC TP53 database: Kato S et al. Proc. Natl. Acad. Sci. USA. 2003 Jul;100(14):8424-9; Monti P et al. Mol. Cancer Res. 2011 Mar;9:271-9). Studies conducted in human cell lines indicate this alteration remains proficient at growth suppression (Kotler E et al. Mol.Cell, 2018 Jul;71:178-190.e8; Giacomelli AO et al. Nat. Genet., 2018 Oct;50:1381-1387). Additional studies in human Saos-2 cell lines showed gain of function properties such as upregulation of several promoters, growth in soft agar, and an increase in DNA synthesis (Shi XB et al. Prostate. 2002 Apr;51:59-72). Crystal structural analysis predict this alteration may cause destabilization of the protein (Tu C et al. Acta Crystallogr. D Biol. Crystallogr. 2008 May;64:471-7). This amino acid position is highly conserved in available vertebrate species. In addition, this alteration is predicted to be deleterious by in silico analysis. This alteration has been detected in the literature and numerous times in our laboratory, however, never in a case that meets classic Li-Fraumeni syndorme or Chompret criteria (Ambry internal data). Although this alteration has not been detected in individuals with Li-Fraumeni syndrome, we can not rule out the possibility that it is a low penetrance risk allele. Based on the available evidence, the clinical significance of this variant remains unclear.
This missense variant replaces arginine with glutamine at codon 282 in the DNA binding domain of the TP53 protein. Computational prediction suggests that this variant may have deleterious impact on protein structure and function (internally defined REVEL score threshold >= 0.7, PMID: 27666373). Functional studies have shown no or partially disruptive effect of this variant on transactivation activity in yeast-based assays (PMID: 11429705, 11896595, 11920959, 12826609, 12909720, 12917626, 21343334) and no effect on the anti-proliferative function of TP53 protein in mammalian cell-based assays (PMID: 29979965, 30224644). This variant has been reported in an infant affected with neuroblastoma with history of lymphoma in her maternal aunt (PMID 10864200). This variant has been observed in an individual affected with lung cancer in his seventies, with family history of breast and prostate cancer in his siblings (PMID: 26787237). This variant has also been observed in individuals affected with breast cancer (PMID: 26976419) and glioneuronal tumor (PMID: 29058119), as well as in an individual affected with colorectal cancer with early-onset breast cancer history in her mother and maternal grandmother (PMID: 29324801). This variant has been identified in 1/251444 chromosomes in the general population by the Genome Aggregation Database (gnomAD). Different variants affecting the same amino acid position (p.Arg282Trp, p.Arg282Pro) are considered to be disease-causing (ClinVar variation ID: 12364, 376659), suggesting that arginine at this position is important for protein function. However, the available clinical and functional evidence is insufficient to determine the role of the p.Arg282Gln variant in disease conclusively. Therefore, this variant is classified as a Variant of Uncertain Significance.
"This variant has been reported in ClinVar as Likely pathogenic (2 clinical laboratories) and as Uncertain significance (11 clinical laboratories) and as Uncertain Significance (1 clinical laboratories)."
COSMIC Somatic Evidence
Open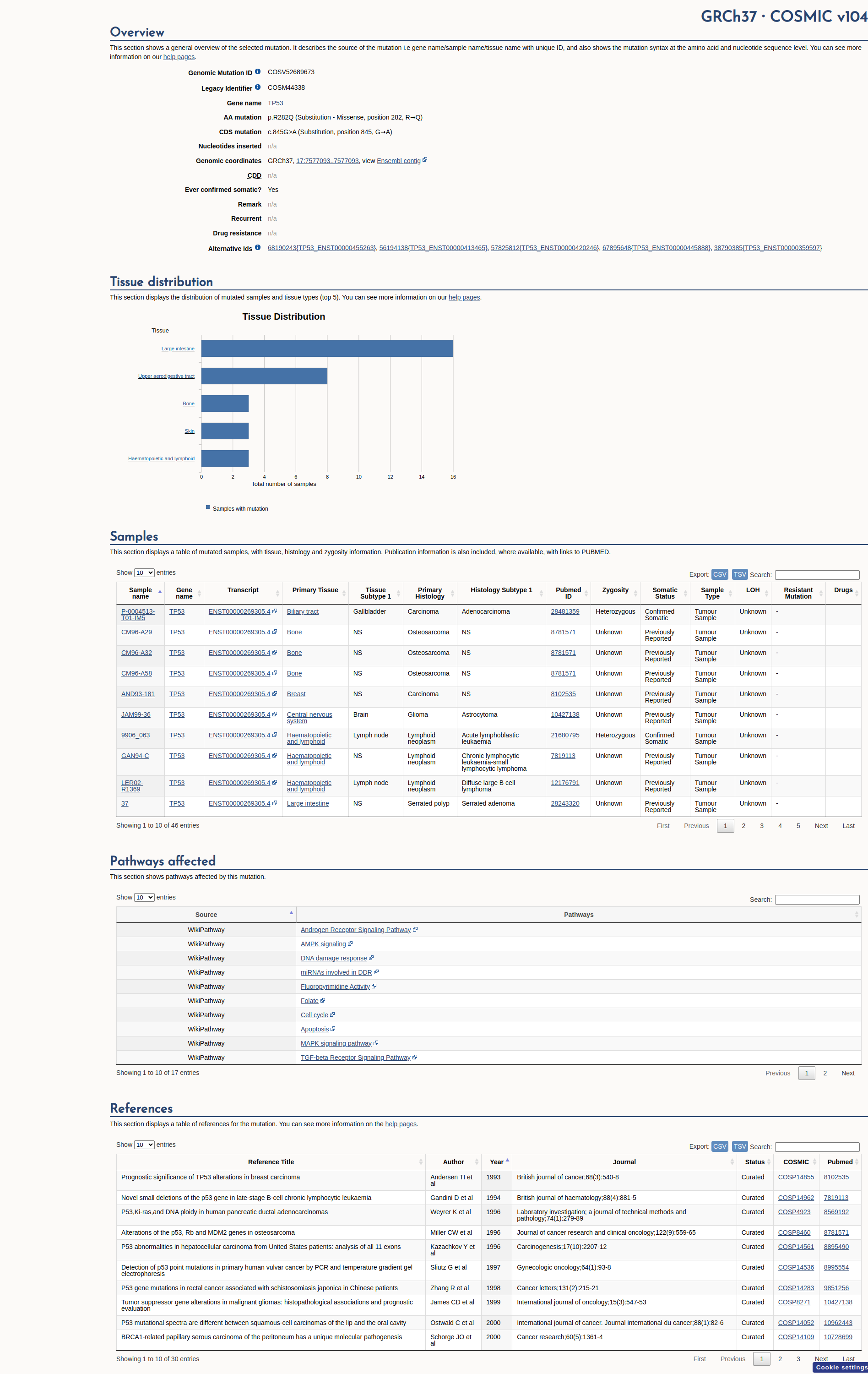
Functional Impact & Domains
Functional Domain
The TP53 R282Q variant has been functionally characterized and is likely damaging. Structural studies indicate that the mutation inactivates the tumor suppressor functions of p53 by altering the flexibility and conformation of the DNA-binding loops. Additionally, functional assays demonstrate a gain of function, as the variant activates PCNA and MDR-1 expression, increases IL-6 levels, and enhances the growth of human cells in culture.
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.01 | 65 bp |
| Donor Loss (DL) | 0.0 | 32 bp |
| Acceptor Gain (AG) | 0.0 | -2 bp |
| Donor Gain (DG) | 0.0 | 158 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, PVS1 applies only to null variants predicted to undergo NMD or to critical splice site changes. This variant is a missense change. Therefore, PVS1 is not applied.
PS1 (Not Applied)
According to VCEP guidelines, PS1_Strong applies to a different nucleotide change resulting in the same amino acid change as a known pathogenic variant. No other variant results in R282Q. Therefore, PS1 is not applied.
PS2 (Not Applied)
According to VCEP guidelines, PS2 applies to confirmed de novo cases with parental testing. No de novo evidence is available. Therefore, PS2 is not applied.
PS3 (Not Applied)
According to VCEP guidelines, PS3 requires non‐functional results on Kato et al. data AND LOF on another assay (Strong) or specified combinations of Kato/Giacomelli/Kotler/Kawaguchi assays. These specific assay data are not provided. Therefore, PS3 is not applied.
PS4 (Not Applied)
According to VCEP guidelines, PS4 applies based on proband case counts reaching point thresholds. No case‐control or proband data are provided. Therefore, PS4 is not applied.
PM1 (Moderate)
According to VCEP guidelines, PM1_Moderate: "Missense variants within the following codons ... 282." The variant affects codon 282, a TP53 hotspot. Therefore, PM1 is applied at Moderate strength.
PM2 (Supporting)
According to VCEP guidelines, PM2_Supporting: "Variant should have an allele frequency of less than 0.00003 (0.003%) in gnomAD ..." The variant has overall MAF=0.000398% (0.00000398), below the threshold when excluding founder populations. Therefore, PM2 is applied at Supporting strength.
PM3 (Not Applied)
According to standard ACMG guidelines, PM3 applies to variants in trans with a pathogenic variant in recessive genes. TP53 is autosomal dominant. Therefore, PM3 is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 applies to protein length changes due to in‐frame indels or stop‐lost. This variant is a missense change. Therefore, PM4 is not applied.
PM5 (Moderate)
According to VCEP guidelines, PM5_Moderate: "Missense variant at an amino acid residue where 1 different missense variant previously determined to be pathogenic ..." R282W is a known TP53 pathogenic variant at the same residue. Therefore, PM5 is applied at Moderate strength.
PM6 (Not Applied)
According to VCEP guidelines, PM6 applies to presumed de novo without confirmation. No such data are provided. Therefore, PM6 is not applied.
PP1 (Not Applied)
According to VCEP guidelines, PP1 requires cosegregation in affected family members. No segregation data are provided. Therefore, PP1 is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, PP2 applies to missense variants in genes with low rate of benign missense variation. TP53 has many pathogenic missense variants and no gene‐specific PP2 recommendation. Therefore, PP2 is not applied.
PP3 (Not Applied)
According to VCEP guidelines, PP3 applies when BayesDel ≥0.16 or aGVGD C65 plus no splicing impact. Computational evidence is mixed and BayesDel data are not provided; SpliceAI indicates no splicing impact. Therefore, PP3 is not applied.
PP4 (Not Applied)
According to VCEP guidelines, PP4 applies to variant observations in somatic VAF contexts. Not relevant for germline TP53 variant. Therefore, PP4 is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, PP5 requires a reputable source classification of pathogenicity without available evidence. ClinVar shows conflicting interpretations (Likely Pathogenic vs Uncertain). Therefore, PP5 is not applied.
BA1 (Not Applied)
According to VCEP guidelines, BA1 applies at FAF ≥0.001 in a non-founder ancestry. The variant's overall frequency is far below this threshold. Therefore, BA1 is not applied.
BS1 (Not Applied)
According to VCEP guidelines, BS1 applies at FAF ≥0.0003 but <0.001 in a non-founder ancestry. The variant's frequency is below 0.0003. Therefore, BS1 is not applied.
BS2 (Not Applied)
According to VCEP guidelines, BS2 requires unaffected older females in a single cohort. No such data are provided. Therefore, BS2 is not applied.
BS3 (Not Applied)
According to VCEP guidelines, BS3 requires functional assays showing no LOF on Kato and another assay. The provided functional evidence indicates damaging effects, not normal function. Therefore, BS3 is not applied.
BS4 (Not Applied)
According to VCEP guidelines, BS4 requires lack of segregation in affected family members. No family data are provided. Therefore, BS4 is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 applies to missense variants in genes where only truncating variants cause disease. TP53 disease mechanism is dominant‐negative missense, so BP1 is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 applies when observed in trans with a pathogenic variant in a dominant gene. No such evidence is provided. Therefore, BP2 is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 applies to in-frame repeats or repetitive regions. This variant is a single‐nucleotide missense. Therefore, BP3 is not applied.
BP4 (Not Applied)
According to VCEP guidelines, BP4_Moderate requires BayesDel ≤ -0.008 and SpliceAI < 0.2. BayesDel data are not available. Although SpliceAI indicates no splicing impact, BayesDel cannot be confirmed. Therefore, BP4 is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 applies when variant found in a case with an alternate molecular basis for disease. No such data are provided. Therefore, BP5 is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 applies to a reputable source classification of benign without evidence. No such reports exist. Therefore, BP6 is not applied.
BP7 (Not Applied)
According to VCEP guidelines, BP7 applies to synonymous or intronic variants with no splicing impact. This is a missense variant. Therefore, BP7 is not applied.