NRAS c.35G>A, p.Gly12Asp
NM_002524.4:c.35G>A
COSMIC ID: COSM564
Pathogenic
NRAS p.G12D is classified as Pathogenic based on VCEP‐moderate PS3 (functional gain‐of‐function assays), VCEP‐moderate PM1 (hotspot P-loop), VCEP‐moderate PM5 (other pathogenic changes at codon 12), and supporting evidence PM2, PP3, and PP5.
ACMG/AMP Criteria Applied
PS3
PM2
PM5
PP3
PP5
Genetic Information
Gene & Transcript Details
Gene
NRAS
Transcript
NM_002524.5
MANE Select
Total Exons
7
Strand
Reverse (−)
Reference Sequence
NC_000001.10
Alternative Transcripts
| ID | Status | Details |
|---|---|---|
| NM_002524.3 | Alternative | 7 exons | Reverse |
| NM_002524.2 | Alternative | 7 exons | Reverse |
| NM_002524.4 | Alternative | 7 exons | Reverse |
Variant Details
HGVS Notation
NM_002524.4:c.35G>A
Protein Change
G12D
Location
Exon 2
(Exon 2 of 7)
5'Exon Structure (7 total)3'
Functional Consequence
Loss of Function
Related Variants
ClinVar reports other pathogenic variants at position 12: G12F, G12A, G12V, G12S, G12R, G12C
Alternate Identifiers
COSM564
Variant interpretation based on transcript NM_002524.5
Genome Browser
Loading genome browser...
HGVS InputNM_002524:c.35G>A
Active Tracks
ConservationRefSeqClinVargnomAD
Navigation tips: Use mouse to drag and zoom. Click on features for details.
Clinical Data
Global Frequency
0.000795%
Very Rare
Highest in Population
European (Finnish)
0.00462%
Rare
Global: 0.000795%
European (Finnish): 0.00462%
0%
0.05%
0.1%
1%
5%
10%+
Allele Information
Total: 251484Alt: 2Homozygotes: 0
ACMG Criteria Applied
PM2
This variant is present in gnomAD (MAF= 0.000795%, 2/251484 alleles, homozygotes = 0) and at a higher frequency in the European (Finnish) population (MAF= 0.00462%, 1/21648 alleles, homozygotes = 0). The variant is rare (MAF < 0.1%), supporting PM2 criterion application.
Classification
5 publications
Likely Pathogenic
Based on 13 submitter reviews in ClinVar
Submitter Breakdown
11 Path
2 LP
Pathogenic
Likely Path.
VUS
Likely Benign
Benign
Publications (5)
A Heterozygous Missense variant c.35G>A in Exon 2 of the NRAS gene that results in the amino acid substitution p.Gly12Asp was identified. The observed variant is novel in gnomAD exomes and genomes, respectively. The severity of the impact of this variant on the protein is medium, based on the effect of the protein and REVEL score. Rare Exome Variant Ensemble Learner (REVEL) is an ensembl method for predicting the pathogenicity of missense variants based on a combination of scores from 13 individual tools: MutPred, FATHMM v2.3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP++, SiPhy, phyloP, and phastCons. The REVEL score for an individual missense variant can range from 0 to 1, with higher scores reflecting greater likelihood that the variant is disease-causing. ClinVar has also classified this variant as Pathogenic/Likely Pathogenic [Variation ID: 39648]. The observed variation has been previously reported for primary melanoma of the CNS in children [Pedersen M, et.al, 2013]. Published functional studies suggests that the variant promotes oncogenesis/leukemogenesis [Wang et al., 2011]. For these reasons, this variant has been classified as Pathogenic.
The p.G12D pathogenic mutation (also known as c.35G>A), located in coding exon 1 of the NRAS gene, results from a G to A substitution at nucleotide position 35. The glycine at codon 12 is replaced by aspartic acid, an amino acid with similar properties. This mutation has been detected in various types of cancers (van 't Veer LJ et al. Mol Cell Biol, 1989 Jul;9:3114-6; Vogelstein B et al. Genes Chromosomes Cancer, 1990 Jul;2:159-62; Brose MS et al. Cancer Res, 2002 Dec;62:6997-7000; Bacher U et al. Blood, 2006 May;107:3847-53; Matsuda K et al. Blood, 2007 Jun;109:5477-80; Vaughn CP et al. Genes Chromosomes Cancer, 2011 May;50:307-12). This variant has also been reported to be de novo in an individual with features consistent with Noonan syndrome (Altmüller F et al. Eur J Hum Genet, 2017 Jun;25:823-831). In addition, in vitro functional studies demonstrated that the mutation increases downstream Ras signaling (Tyner JW et al. Blood, 2009 Feb;113:1749-55). Embryonic expression of this mutation led to embryonic lethality and cardiac developmental defects (You X et al. Front Cell Dev Biol, 2021 Feb;9:633661). Endogenous expression of this variant at weaning induced myeloproliferative disorder, and mice that died of a spectrum of hematologic malignancies (Li Q et al. Blood, 2011 Feb;117:2022-32). This amino acid position is highly conserved in available vertebrate species. In addition, this alteration is predicted to be deleterious by in silico analysis. Based on the supporting evidence, this alteration is interpreted as a disease-causing mutation.
This sequence change replaces glycine, which is neutral and non-polar, with aspartic acid, which is acidic and polar, at codon 12 of the NRAS protein (p.Gly12Asp). The frequency data for this variant in the population databases is considered unreliable, as metrics indicate poor data quality at this position in the gnomAD database. This missense change has been observed in individual(s) with Noonan syndrome (PMID: 28594414). In at least one individual the variant was observed to be de novo. ClinVar contains an entry for this variant (Variation ID: 39648). Advanced modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) performed at Invitae indicates that this missense variant is expected to disrupt NRAS protein function with a positive predictive value of 95%. This variant disrupts the p.Gly12 amino acid residue in NRAS. Other variant(s) that disrupt this residue have been determined to be pathogenic (PMID: 28098151, 28594414). This suggests that this residue is clinically significant, and that variants that disrupt this residue are likely to be disease-causing. For these reasons, this variant has been classified as Pathogenic.
The variant is observed at an extremely low frequency in the gnomAD v2.1.1 dataset (total allele frequency: <0.001%). Predicted Consequence/Location: Missense changes are a common disease-causing mechanism. In silico tool predictions suggest damaging effect of the variant on gene or gene product (REVEL: 0.78; 3Cnet: 0.96). Same nucleotide change resulting in same amino acid change (ClinVar ID: VCV000039648 /PMID: 28594414 /3billion dataset) and different missense changes at the same codon (p.Gly12Ala, p.Gly12Arg, p.Gly12Cys, p.Gly12Pro, p.Gly12Ser, p.Gly12Val / ClinVar ID: VCV000040468, VCV000040469, VCV000040470, VCV000177778, VCV000219097 /PMID: 23334668, 28594414, 30417923, 32888943 /3billion dataset) have been previously reported as pathogenic/likely pathogenic with strong evidence.The variant has been previously reported as assumed (i.e. paternity and maternity not confirmed) de novo in at least one similarly affected unrelated individual (3billion dataset). Therefore, this variant is classified as Pathogenic according to the recommendation of ACMG/AMP guideline.
Clinical Statement
This variant has been reported in ClinVar as Pathogenic (11 clinical laboratories) and as Likely pathogenic (2 clinical laboratories).
COSMIC ID
COSM564
Recurrence
1158 occurrences
PM1 Criteria
Applied
Criterion PM1 is applied based on the high recurrence in COSMIC database.
COSMIC Database Preview
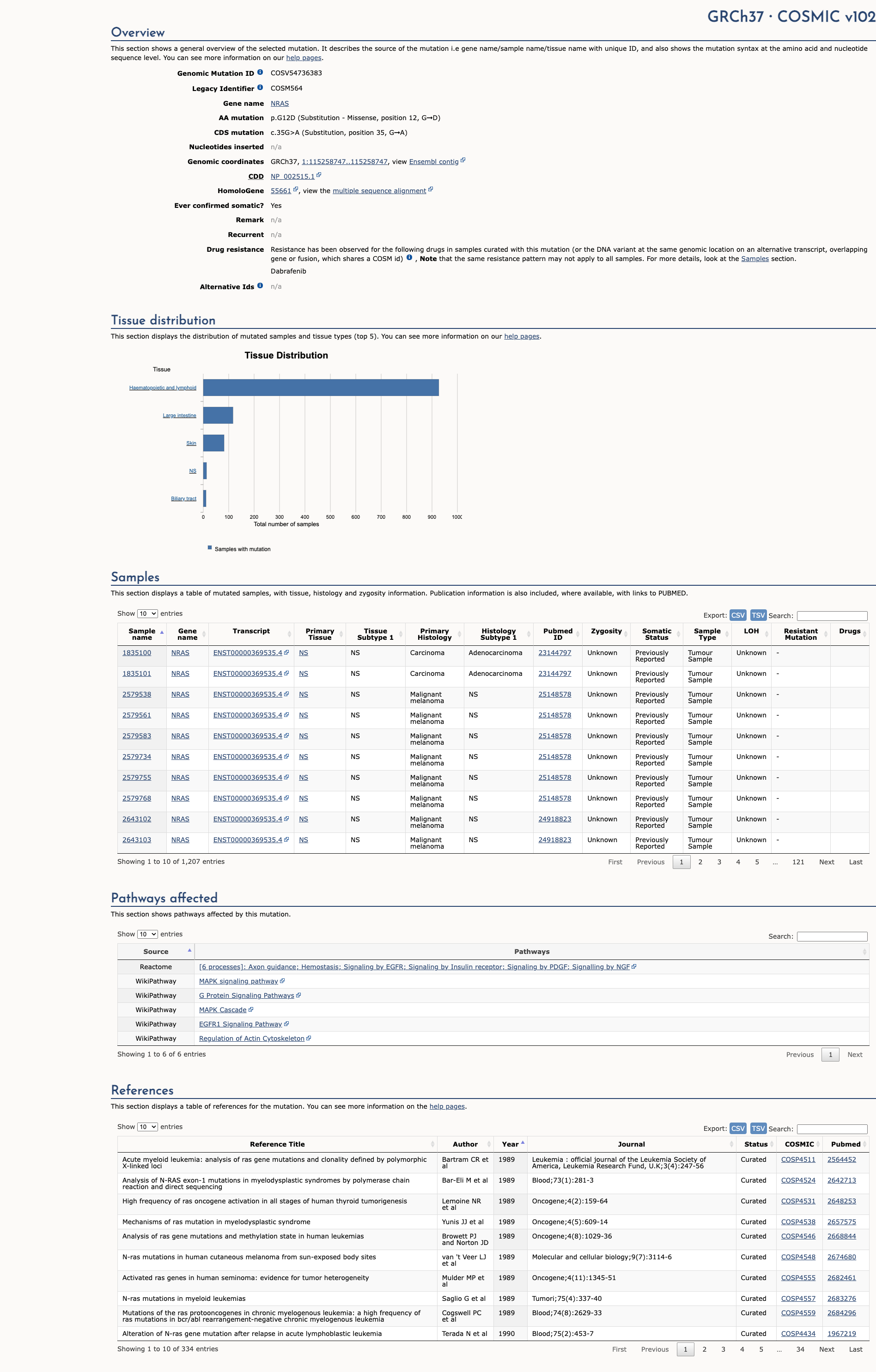
Accessing full COSMIC database details requires institutional login or subscription. External links may prompt for authentication.
Functional Impact
Functional Domain
Hotspot Status
Hotspot
PM1
Mutation Count
444
Reported mutations in this domain
050100+
Domain Summary
This variant is located in a mutational hotspot or critical domain (444 mutations).
PM1 criterion applied.
Related Variants in This Domain
ClinVar reports other pathogenic variants at position 12: G12F, G12A, G12V, G12S, G12R, G12C
PM5 criterion applied.
Functional Summary
Gain-of-Function
The NRAS G12D variant has been functionally characterized as a gain-of-function mutation. It is located in the GTP-binding domain of the NRAS protein and results in decreased intrinsic GTPase activity and reduced sensitivity to GTPase-activating proteins. This leads to the accumulation of GTP-bound NRAS, activation of downstream signaling pathways, increased cell proliferation, and leukemogenesis in mouse models. Additionally, structural analysis indicates that mutations at the G12 position render the protein constitutively active. These findings support the conclusion that NRAS G12D is an activating and oncogenic mutation.
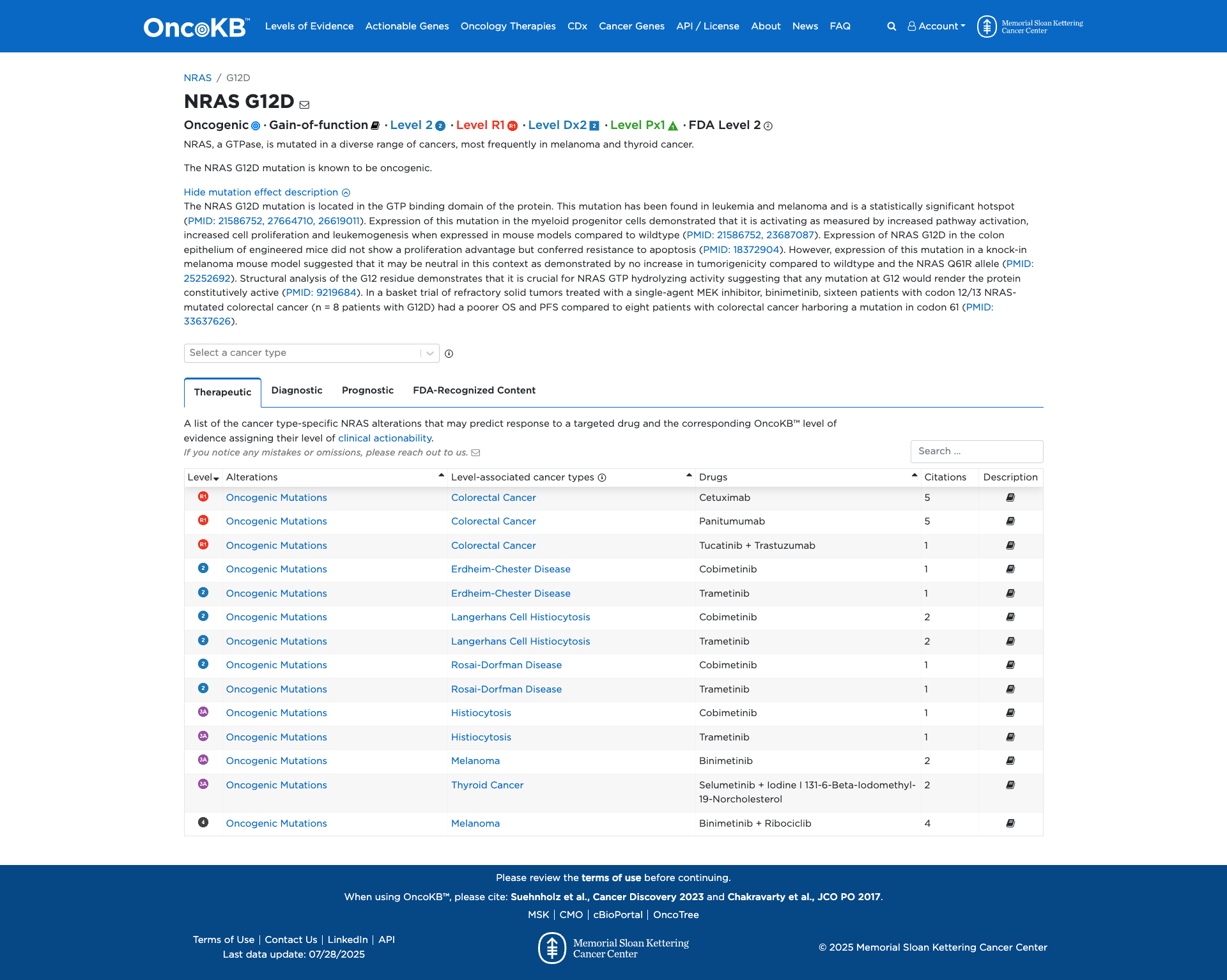
Database Previews
OncoKB
JAX-CKB
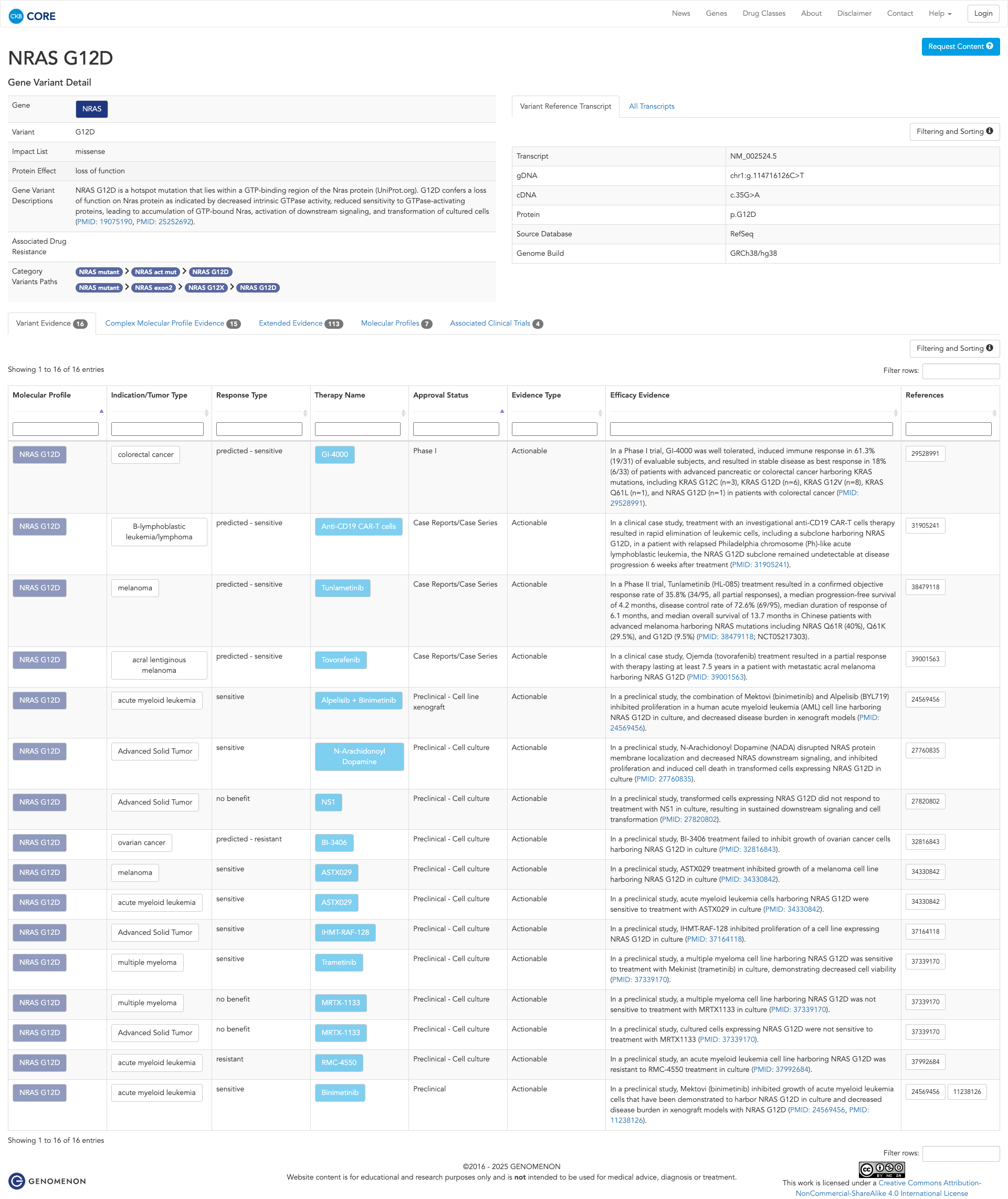
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
REVEL Score
0.783
0.783
Likely Benign0.0
Uncertain (Low)0.2
Uncertain (Med)0.5
Likely Pathogenic0.75
REVEL scores ≥ 0.75 are strong evidence (PP3)
Predictor Consensus
Mixed/VUS
PP3 Applied
Yes
Additional Predictors
Benign:
CADD: 5.51
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific) VCEP Guidelines
PVS1
PVS1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PVS1 is: 'Very Strong null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a gene where loss of function is a known mechanism of disease.' The evidence for this variant shows: it is a missense change (G12D) in NRAS, a gain-of-function oncogene. Therefore, this criterion is not applied because the variant is not a null variant and LOF is not the mechanism.
PS1
PS1 (Not Applied) Strength Modified
According to VCEP guidelines, the rule for PS1 is: 'Strong Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. Applicable for observed analogous pathogenic residue positions in HRAS, KRAS, MRAS, NRAS, RIT1, and RRAS2.' The evidence for this variant shows: G12D has been observed via the same nucleotide change (c.35G>A) and no alternate nucleotide change yielding G12D has been reported. Therefore, this criterion is not applied because there is no distinct nucleotide change producing the same amino acid change.
PS2
PS2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS2 is: 'Very Strong de novo (both maternity and paternity confirmed) in a patient with the disease and no family history.' The evidence for this variant shows: no de novo data are available. Therefore, this criterion is not applied due to absence of de novo confirmation.
PS3
PS3 (Moderate) Strength Modified
According to VCEP guidelines, the rule for PS3 is: 'Moderate Strength: Moderate Two or more different approved assays.' The evidence for this variant shows: multiple in vitro and in vivo functional studies demonstrate gain-of-function (decreased GTPase activity, reduced sensitivity to GAPs, accumulation of GTP-bound NRAS, leukemogenesis in mouse models). Therefore, this criterion is applied at Moderate strength because two or more different approved assays support a damaging effect.
PS4
PS4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS4 is: 'Strong The prevalence of the variant in affected individuals is significantly increased compared to controls.' The evidence for this variant shows: no case-control or proband counting data are provided. Therefore, this criterion is not applied due to lack of case prevalence data.
PM1
PM1 (Moderate)
According to VCEP guidelines, the rule for PM1 is: 'Moderate Applicable only to critical and well-established functional domains available in the supplementary table (P-loop [AA 10-17], SW1 [AA 25-40], SW2 [AA 57-64], SAK [AA 145-156]). Not applicable to specific amino acid residues (see PM5).' The evidence for this variant shows: G12D is located in the P-loop (AA 10–17), a critical GTP-binding domain without benign variation. Therefore, this criterion is applied at Moderate strength because the variant lies in a well-established functional domain.
PM2
PM2 (Supporting) Strength Modified
According to VCEP guidelines, the rule for PM2 is: 'Supporting The variant must be absent from controls (gnomAD).' The evidence for this variant shows: extremely rare in gnomAD (MAF = 0.000795%). Therefore, this criterion is applied at Supporting strength because the variant is absent or extremely rare in population controls.
PM3
PM3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM3 is: 'Moderate For recessive disorders, detected in trans with a pathogenic variant.' The evidence for this variant shows: no evidence of trans configuration with another variant. Therefore, this criterion is not applied due to lack of trans data.
PM4
PM4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM4 is: 'Moderate Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants.' The evidence for this variant shows: it is a missense change without any indel or protein length alteration. Therefore, this criterion is not applied.
PM5
PM5 (Moderate)
According to VCEP guidelines, the rule for PM5 is: 'Moderate 1 [likely] pathogenic residue change at the same codon.' The evidence for this variant shows: other pathogenic missense variants (e.g., G12C, G12V, G12R, G12S) have been reported at codon 12. Therefore, this criterion is applied at Moderate strength because a different pathogenic residue change at the same codon has been seen.
PM6
PM6 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM6 is: 'Moderate Assumed de novo, but without confirmation of paternity and maternity.' The evidence for this variant shows: no de novo or parental data. Therefore, this criterion is not applied.
PP1
PP1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP1 is: 'Supporting Co-segregation with disease in multiple affected family members.' The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied due to lack of segregation information.
PP2
PP2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP2 is: 'Supporting Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease.' The evidence for this variant shows: NRAS is known for gain-of-function missense hotspots, but specific quantitative data on benign missense rate are not provided. Therefore, this criterion is not applied.
PP3
PP3 (Supporting)
According to VCEP guidelines, the rule for PP3 is: 'Supporting For missense variants: REVEL ≥ 0.7.' The evidence for this variant shows: REVEL score = 0.78, exceeding the threshold. Therefore, this criterion is applied at Supporting strength because computational evidence supports a deleterious effect.
PP4
PP4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP4 is: 'Supporting Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology.' The evidence for this variant shows: no phenotype or family history data. Therefore, this criterion is not applied.
PP5
PP5 (Supporting)
According to standard ACMG guidelines, the rule for PP5 is: 'Supporting Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation.' The evidence for this variant shows: ClinVar submissions list it as Pathogenic (11 labs) and Likely Pathogenic (2 labs). Therefore, this criterion is applied at Supporting strength because of multiple reputable assertions.
BA1
BA1 (Not Applied) Strength Modified
According to VCEP guidelines, the rule for BA1 is: 'Stand Alone GnomAD filtering allele frequency ≥ 0.05%.' The evidence for this variant shows: MAF = 0.000795% < 0.05%. Therefore, this criterion is not applied.
BS1
BS1 (Not Applied) Strength Modified
According to VCEP guidelines, the rule for BS1 is: 'Strong GnomAD filtering allele frequency ≥ 0.025%.' The evidence for this variant shows: MAF = 0.000795% < 0.025%. Therefore, this criterion is not applied.
BS2
BS2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS2 is: 'Strong Observed in a healthy adult with full penetrance at an early age.' The evidence for this variant shows: no data on occurrence in healthy individuals. Therefore, this criterion is not applied.
BS3
BS3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS3 is: 'Strong Well-established functional studies show no damaging effect.' The evidence for this variant shows: functional studies indicate gain-of-function damage. Therefore, this criterion is not applied.
BS4
BS4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS4 is: 'Strong Lack of segregation in affected members of a family.' The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1
BP1 (Not Applied) Strength Modified
According to VCEP guidelines, the rule for BP1 is: 'Supporting This rule has contraindications for RASopathies; used for truncating variants when LOF is not disease mechanism.' The evidence for this variant shows: it is a missense variant in NRAS, a gain-of-function gene. Therefore, this criterion is not applied.
BP2
BP2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP2 is: 'Supporting Observed in trans with a pathogenic variant for a fully penetrant dominant disorder or cis with a pathogenic variant.' The evidence for this variant shows: no data on cis/trans configuration. Therefore, this criterion is not applied.
BP3
BP3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP3 is: 'Supporting In-frame deletions/insertions in a repetitive region without a known function.' The evidence for this variant shows: it is a missense change, not in-frame indel. Therefore, this criterion is not applied.
BP4
BP4 (Not Applied) Strength Modified
According to VCEP guidelines, the rule for BP4 is: 'Supporting For missense variants: REVEL ≤ 0.3.' The evidence for this variant shows: REVEL = 0.78 > 0.3. Therefore, this criterion is not applied.
BP5
BP5 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP5 is: 'Supporting Variant found in a case with an alternate molecular basis for disease.' The evidence for this variant shows: no data on alternate molecular diagnoses. Therefore, this criterion is not applied.
BP6
BP6 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP6 is: 'Supporting Reputable source reports variant as benign but evidence is not available.' The evidence for this variant shows: no benign assertions in reputable databases. Therefore, this criterion is not applied.
BP7
BP7 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP7 is: 'Supporting Synonymous variant with no predicted splice impact and no conservation.' The evidence for this variant shows: it is a missense change. Therefore, this criterion is not applied.