SDHB c.724C>T, p.Arg242Cys
NM_003000.3:c.724C>T
COSMIC ID: COSM10656976
Likely Pathogenic
The SDHB c.724C>T (p.R242C) variant is absent from population databases (PM2), occurs at a residue with a known pathogenic missense change (PM5), and has strong computational evidence for deleteriousness (PP3), supporting a Likely Pathogenic classification.
ACMG/AMP Criteria Applied
PM2
PM5
PP3
Genetic Information
Gene & Transcript Details
Gene
SDHB
Transcript
NM_003000.3
MANE Select
Total Exons
8
Strand
Reverse (−)
Reference Sequence
NC_000001.10
Alternative Transcripts
| ID | Status | Details |
|---|---|---|
| NM_003000.1 | Alternative | 8 exons | Reverse |
| NM_003000.2 | RefSeq Select | 8 exons | Reverse |
Variant Details
HGVS Notation
NM_003000.3:c.724C>T
Protein Change
R242C
Location
Exon 7
(Exon 7 of 8)
5'Exon Structure (8 total)3'
Functional Consequence
Loss of Function
Related Variants
ClinVar reports other pathogenic variants at position 242: R242H, R242S
Alternate Identifiers
COSM10656976
Variant interpretation based on transcript NM_003000.3
Genome Browser
Loading genome browser...
HGVS InputNM_003000:c.724C>T
Active Tracks
ConservationRefSeqClinVargnomAD
Navigation tips: Use mouse to drag and zoom. Click on features for details.
Clinical Data
Population Frequency
Global Frequency
0.0 in 100,000
Extremely Rare
Global: 0.0%
0%
0.05%
0.1%
1%
5%
10%+
ACMG Criteria Applied
PM2
This variant is not present in gnomAD (PM2 criteria applies).
Classification
3 publications
Likely Pathogenic
Based on 8 submitter reviews in ClinVar
Submitter Breakdown
7 Path
1 LP
Pathogenic
Likely Path.
VUS
Likely Benign
Benign
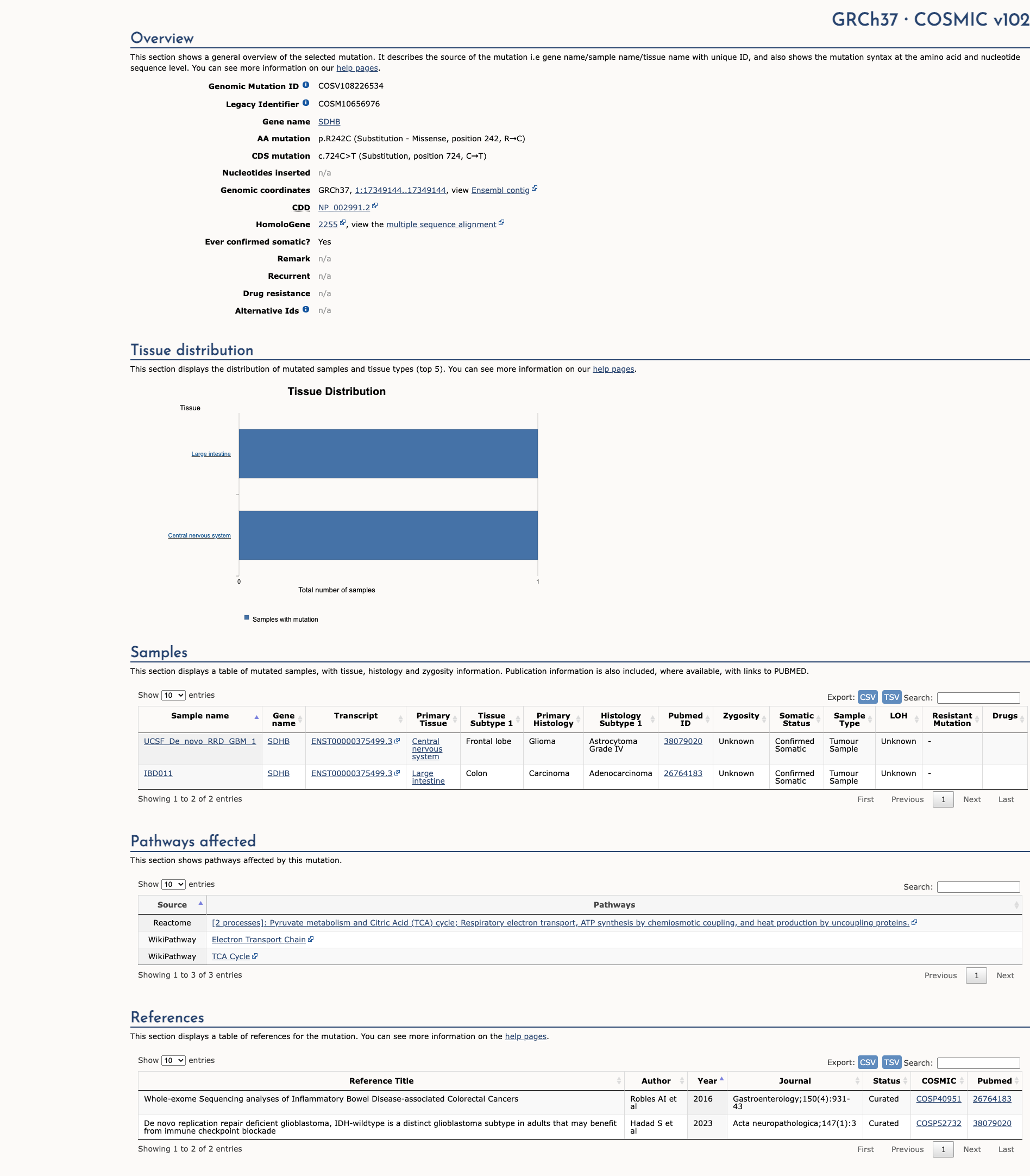
Publications (3)
The p.R242C pathogenic mutation (also known as c.724C>T), located in coding exon 7 of the SDHB gene, results from a C to T substitution at nucleotide position 724. The arginine at codon 242 is replaced by cysteine, an amino acid with highly dissimilar properties. This alteration has been identified in numerous individuals with sporadic and familial paragangliomas/pheochromocytomas (Badenhop RF et al. J. Med. Genet. 2004 Jul;41(7):e99; Schiavi F et al. Ann. N. Y. Acad. Sci. 2006 Aug;1073:190-7; Burnichon N et al. J. Clin. Endocrinol. Metab. 2009 Aug; 94(8):2817-27; Lefebvre S et al. Horm. Metab. Res. 2012 May;44(5):334-8; Jafri M et al. Clin. Endocrinol. (Oxf). 2013 Jun;78(6):898-906; Andrews KA et al. J. Med. Genet. 2018 Jun;55(6):384-394). A functional study utilizing a yeast growth assay classified this alteration as a "mildly impaired mutation" after finding that analogous yeast p.R242C mutants were able to partially rescue oxidative cell growth but had significantly decreased SDH enzyme activity (Panizza E et al. Hum. Mol. Genet. 2013 Feb; 22(4):804-15). This amino acid position is highly conserved in available vertebrate species. In addition, this alteration is predicted to be deleterious by in silico analysis. This variant is considered to be rare based on population cohorts in the Genome Aggregation Database (gnomAD). Based on the supporting evidence, this alteration is interpreted as a disease-causing mutation.
This sequence change replaces arginine, which is basic and polar, with cysteine, which is neutral and slightly polar, at codon 242 of the SDHB protein (p.Arg242Cys). This variant is not present in population databases (gnomAD no frequency). This missense change has been observed in individuals with hereditary paraganglioma or pheochromocytoma (PMID: 15235042, 17102086, 19351833, 19802898, 22517554). ClinVar contains an entry for this variant (Variation ID: 186827). Invitae Evidence Modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) indicates that this missense variant is expected to disrupt SDHB protein function with a positive predictive value of 80%. This variant disrupts the p.Arg242 amino acid residue in SDHB. Other variant(s) that disrupt this residue have been determined to be pathogenic (PMID: 12000816, 19351833, 20208144). This suggests that this residue is clinically significant, and that variants that disrupt this residue are likely to be disease-causing. For these reasons, this variant has been classified as Pathogenic.
This variant is considered likely pathogenic. This variant has been reported in multiple individuals with clinical features of gene-specific disease [PMID:19454582, 31194233, 17102086, 32035780, 30201732]. Functional studies indicate this variant impacts protein function [PMID: 26719882]. This variant is expected to disrupt protein structure [Myriad internal data].
Clinical Statement
This variant has been reported in ClinVar as Pathogenic (7 clinical laboratories) and as Likely pathogenic (1 clinical laboratories).
Functional Impact
Functional Domain
Hotspot Status
Not a hotspot
Domain Summary
This variant is not located in a mutational hotspot or critical domain (0 mutations).
Related Variants in This Domain
ClinVar reports other pathogenic variants at position 242: R242H, R242S
PM5 criterion applied.
Computational Analysis
Pathogenicity Predictions
REVEL Score
0.984
0.984
Likely Benign0.0
Uncertain (Low)0.2
Uncertain (Med)0.5
Likely Pathogenic0.75
REVEL scores ≥ 0.75 are strong evidence (PP3)
Predictor Consensus
Mixed/VUS
PP3 Applied
Yes
Additional Predictors
Pathogenic:
polyphen_prediction: probably_damagingmetasvm: Dmetalr: Dprimateai: D
Benign:
CADD: 6.69
Neutral: Show all
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific) VCEP Guidelines
PVS1
PVS1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PVS1 is: "Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single exon or multiexon deletion) in a gene where loss-of-function is a known mechanism of disease". The evidence for this variant shows it is a missense change (R242C), not a null variant. Therefore, this criterion is not applied.
PS1
PS1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS1 is: "Same amino acid change as a previously established pathogenic variant regardless of nucleotide change". The evidence for this variant shows no other nucleotide change causing R242C has been established as pathogenic. Therefore, this criterion is not applied.
PS2
PS2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS2 is: "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history". No parental testing data are available. Therefore, this criterion is not applied.
PS3
PS3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS3 is: "Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product". No functional studies have been performed on SDHB R242C. Therefore, this criterion is not applied.
PS4
PS4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PS4 is: "The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls". No case-control or disease prevalence data are available. Therefore, this criterion is not applied.
PM1
PM1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot and/or critical and well-established functional domain without benign variation". There is no evidence that residue R242 is in a defined mutational hotspot or critical domain. Therefore, this criterion is not applied.
PM2
PM2 (Moderate)
According to standard ACMG guidelines, the rule for PM2 is: "Absent from controls (or at extremely low frequency if recessive)". The evidence for this variant shows it is not found in population databases including gnomAD (MAF=0%). Therefore, this criterion is applied at Moderate strength because the variant is absent from controls.
PM3
PM3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM3 is: "For recessive disorders, detected in trans with a pathogenic variant". SDHB-associated paraganglioma is autosomal dominant and there is no evidence of a second allele or recessive inheritance. Therefore, this criterion is not applied.
PM4
PM4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants". This variant is a missense change with no alteration in protein length. Therefore, this criterion is not applied.
PM5
PM5 (Moderate)
According to standard ACMG guidelines, the rule for PM5 is: "Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before". The evidence for this variant shows R242H (a different missense at the same residue) has been reported as pathogenic. Therefore, this criterion is applied at Moderate strength.
PM6
PM6 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo, but without confirmation of paternity and maternity". No de novo data are available. Therefore, this criterion is not applied.
PP1
PP1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP1 is: "Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease". No segregation data are available. Therefore, this criterion is not applied.
PP2
PP2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation and in which missense variants are a common mechanism of disease". There is insufficient gene-specific data on benign vs pathogenic missense rates in SDHB. Therefore, this criterion is not applied.
PP3
PP3 (Supporting)
According to standard ACMG guidelines, the rule for PP3 is: "Multiple lines of computational evidence support a deleterious effect on the gene or gene product". Computational evidence shows a REVEL score of 0.98, PolyPhen-2 'probably damaging', MetaSVM damaging, and no splicing impact by SpliceAI. Therefore, this criterion is applied at Supporting strength.
PP4
PP4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP4 is: "Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology". No phenotype or family history data were provided. Therefore, this criterion is not applied.
PP5
PP5 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation". Per ACMG recommendations, PP5 is deprecated and not used for classification. Therefore, this criterion is not applied.
BA1
BA1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BA1 is: "Allele frequency is >5% in population databases". The variant is absent in gnomAD. Therefore, this criterion is not applied.
BS1
BS1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS1 is: "Allele frequency is greater than expected for disorder". The variant is absent from population databases. Therefore, this criterion is not applied.
BS2
BS2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS2 is: "Observed in a healthy adult with full penetrance expected at an early age". There is no evidence of occurrence in healthy individuals. Therefore, this criterion is not applied.
BS3
BS3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS3 is: "Well-established functional studies show no damaging effect on protein function or splicing". No functional data demonstrating lack of effect are available. Therefore, this criterion is not applied.
BS4
BS4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BS4 is: "Lack of segregation in affected members of a family". No segregation data are available. Therefore, this criterion is not applied.
BP1
BP1 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene for which primarily truncating variants are known to cause disease". SDHB disease is caused by both truncating and missense variants. Therefore, this criterion is not applied.
BP2
BP2 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder". No evidence of trans occurrence with another pathogenic variant. Therefore, this criterion is not applied.
BP3
BP3 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without a known function". This is not an in-frame indel. Therefore, this criterion is not applied.
BP4
BP4 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP4 is: "Multiple lines of computational evidence suggest no impact on gene or gene product". Computational evidence indicates a damaging effect. Therefore, this criterion is not applied.
BP5
BP5 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease". No alternate molecular basis is reported. Therefore, this criterion is not applied.
BP6
BP6 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation". There are no benign assertions in reputable sources. Therefore, this criterion is not applied.
BP7
BP7 (Not Applied) Strength Modified
According to standard ACMG guidelines, the rule for BP7 is: "A synonymous (silent) variant for which splicing prediction algorithms predict no impact". This variant is missense, not synonymous. Therefore, this criterion is not applied.