Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000251.2 | RefSeq Select | 3226 nt | 126–2930 |
| NM_000251.1 | Alternative | 3145 nt | 69–2873 |
| NM_000251.3 | MANE Select | 3115 nt | 37–2841 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThis sequence change creates a premature translational stop signal (p.Gln324*) in the MSH2 gene. It is expected to result in an absent or disrupted protein product. Loss-of-function variants in MSH2 are known to be pathogenic (PMID: 15849733, 24362816). This variant is not present in population databases (gnomAD no frequency). This premature translational stop signal has been observed in individual(s) with Lynch syndrome (PMID: 16142001). ClinVar contains an entry for this variant (Variation ID: 91261). For these reasons, this variant has been classified as Pathogenic.
The p.Q324* pathogenic mutation (also known as c.970C>T), located in coding exon 6 of the MSH2 gene, results from a C to T substitution at nucleotide position 970. This changes the amino acid from a glutamine to a stop codon within coding exon 6. This mutation has been previously reported in French patients with hereditary non-polyposis colorectal cancer (HNPCC)/Lynch syndrome (Parc Y et al. J. Med. Genet., 2003 Mar;40:208-13; Bonadona V et al. JAMA, 2011 Jun;305:2304-10). This mutation was also seen in patients who met Amsterdam criteria with MSI-H colorectal cancers demonstrating absent staining of MSH2 on immunohistochemistry (Bécouarn Y et al. Gastroenterol. Clin. Biol. 2005;29:667-75; Paraf F et al. Histopathology, 2001 Sep;39:250-8). One woman from a family with urinary tract cancers was also found to have this mutation (Wischhusen JW et al. Cancer Epidemiol Biomarkers Prev, 2020 01;29:193-199). This variant is considered to be rare based on population cohorts in the Genome Aggregation Database (gnomAD). In addition to the clinical data presented in the literature, this alteration is expected to result in loss of function by premature protein truncation or nonsense-mediated mRNA decay. As such, this alteration is interpreted as a disease-causing mutation.
"This variant has been reported in ClinVar as Pathogenic (5 clinical laboratories) and as Pathogenic by International Society for Gastrointestinal Hereditary Tumours (InSiGHT) expert panel."
COSMIC Somatic Evidence
Open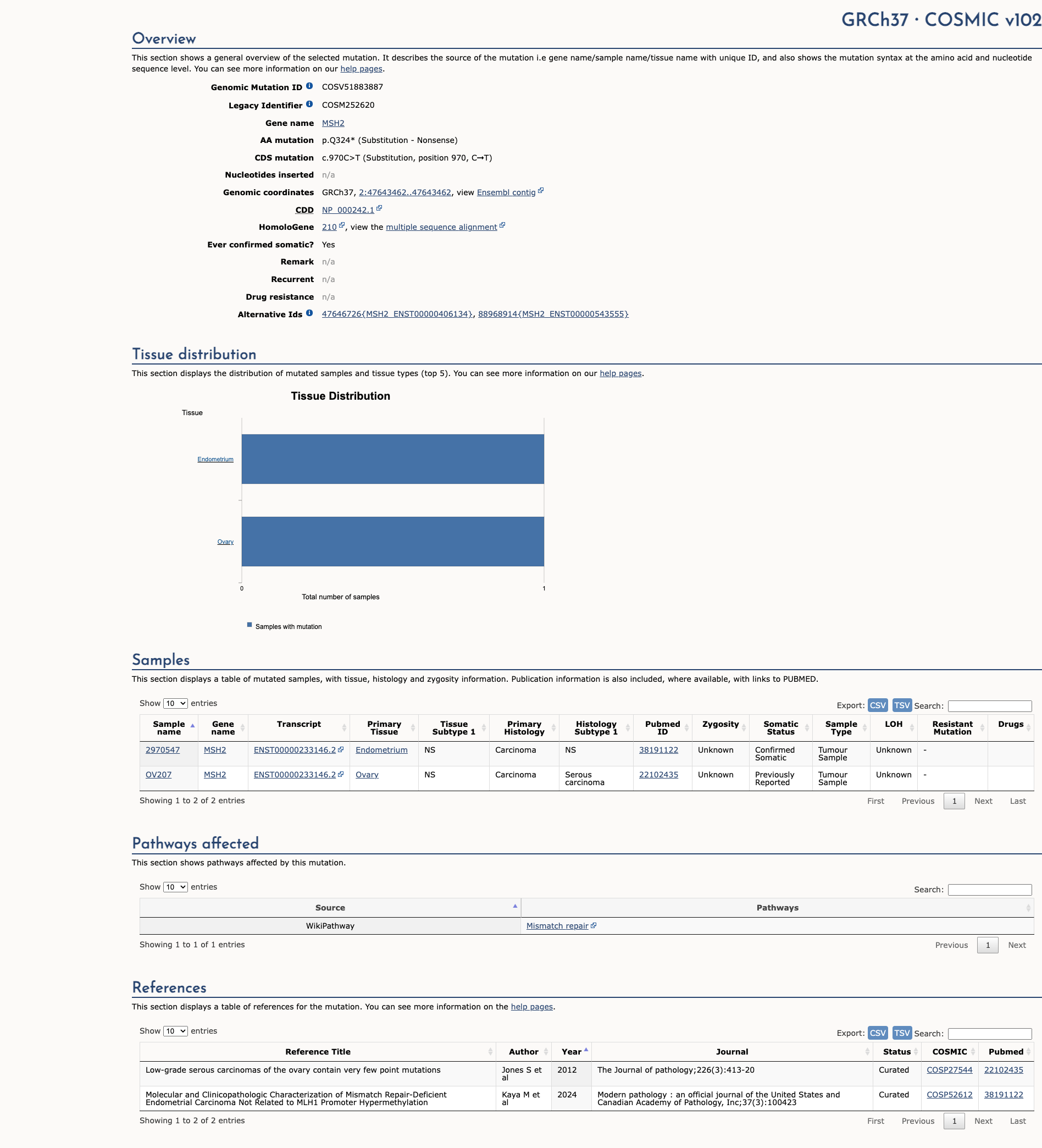
Functional Impact & Domains
Functional Domain
The MSH2 Q324* variant is a truncating mutation that likely results in loss of function of the MSH2 protein, a key component of the DNA mismatch repair pathway. Truncating mutations in MSH2 are associated with Lynch syndrome and lead to impaired mismatch repair due to disruption of essential domains required for nucleotide binding and protein dimerization. This loss of function is consistent with an oncogenic effect, contributing to the development of cancers such as colorectal, small bowel, and endometrial cancers.

Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | -14 bp |
| Donor Loss (DL) | 0.0 | 106 bp |
| Acceptor Gain (AG) | 0.0 | 43 bp |
| Donor Gain (DG) | 0.0 | 110 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to VCEP guidelines for MSH2, the rule for PVS1 is: "Very Strong Nonsense/frameshift variant introducing Premature Termination Codon ≤ codon 891 in MSH2." The evidence for this variant shows: c.970C>T introduces a stop codon at Q324, which is upstream of codon 891 and is not in the last exon. Therefore, this criterion is applied at Very Strong strength because the variant introduces a premature termination codon predicted to undergo NMD in a gene where loss of function is a known mechanism of disease.
PS1 (Not Applied)
According to standard ACMG guidelines, PS1 applies to a missense variant encoding the same amino acid change as a known pathogenic variant. This is a nonsense variant, so the criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, PS2 applies to de novo variants with parental confirmation. No de novo status data are available for this variant, so the criterion is not applied.
PS3 (Not Applied)
According to VCEP guidelines for MSH2, PS3 requires calibrated functional assays with functional odds for pathogenicity >18.7. The evidence for this variant shows predicted loss of function but no calibrated assay data. Therefore, PS3 is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, PS4 applies to increased prevalence of the variant in affected individuals vs. controls. No case-control or case prevalence data are available, so PS4 is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, PM1 applies to variants in mutational hotspots or critical functional domains. No evidence places this truncating variant in a defined hotspot beyond the general LOF mechanism, so PM1 is not applied.
PM2 (Supporting)
According to VCEP guidelines for MSH2, PM2_Supporting is: "Absent/extremely rare (<1 in 50,000 alleles) in gnomAD v4." The evidence for this variant shows: MAF = 0% in gnomAD. Therefore, PM2 is applied at Supporting strength because the variant is absent from large population databases.
PM3 (Not Applied)
According to standard ACMG guidelines, PM3 applies to recessive disorders with detected trans variants. Lynch syndrome is autosomal dominant and no trans data are relevant, so PM3 is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 applies to protein length changes in nonrepeat regions not covered by PVS1. This truncating variant is already covered by PVS1, so PM4 is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, PM5 applies to novel missense changes at a residue with a known pathogenic missense variant. This is a nonsense variant, so PM5 is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, PM6 applies to presumed de novo variants without confirmation. No de novo status data are available, so PM6 is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, PP1 applies to co-segregation in families. No segregation data are available, so PP1 is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, PP2 applies to missense variants in genes with low rate of benign missense variation. This is a truncating variant, so PP2 is not applied.
PP3 (Not Applied)
According to standard ACMG guidelines, PP3 applies to multiple lines of computational evidence supporting a deleterious effect. As a nonsense variant, computational predictions are not applicable, so PP3 is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, PP4 applies to phenotype specificity in Mendelian disorders. No tumor phenotype or MSI/MMR expression data are provided, so PP4 is not applied.
PP5 (Supporting)
According to standard ACMG guidelines, PP5 is: "Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation." The evidence for this variant shows: reported as Pathogenic by ClinVar (5 labs) and InSiGHT expert panel. Therefore, PP5 is applied at Supporting strength because reputable sources classify it as pathogenic without primary data.
BA1 (Not Applied)
According to VCEP guidelines for MSH2, BA1 applies to allele frequency ≥0.1% in gnomAD. The variant is absent, so BA1 is not applied.
BS1 (Not Applied)
According to VCEP guidelines for MSH2, BS1 applies to allele frequency 0.01–0.1% in gnomAD. The variant is absent, so BS1 is not applied.
BS2 (Not Applied)
According to VCEP guidelines for MSH2, BS2 requires co-occurrence in trans with a known pathogenic variant in a CMMRD-excluded individual. No such data are available, so BS2 is not applied.
BS3 (Not Applied)
According to VCEP guidelines for MSH2, BS3 applies to well-established functional assays showing no damaging effect. No benign functional data are available, so BS3 is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, BS4 applies to lack of segregation in families. No segregation data are available, so BS4 is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 applies to missense variants in a gene where only truncating variants cause disease. This is a truncating variant, so BP1 is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 applies to observed in trans with a pathogenic variant in recessive conditions. Not applicable for this dominant condition, so BP2 is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 applies to in-frame indels in repetitive regions. This is a nonsense variant, so BP3 is not applied.
BP4 (Not Applied)
According to standard ACMG guidelines, BP4 applies to computational evidence supporting benign impact. As a nonsense variant, computational predictions are not sufficient, so BP4 is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 applies when variant found in a case with an alternative molecular basis for disease. No such data are available, so BP5 is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 applies to reputable source reporting benign. No benign classifications exist, so BP6 is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, BP7 applies to synonymous variants with no splicing impact. This is a nonsense variant, so BP7 is not applied.