Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_022552.4 | RefSeq Select | 4324 nt | 268–3006 |
| NM_022552.5 | MANE Select | 9421 nt | 278–3016 |
| NM_022552.3 | Alternative | 4314 nt | 258–2996 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
Open""
COSMIC Somatic Evidence
Open
Functional Impact & Domains
Functional Domain
The DNMT3A P700Hfs*5 variant is a truncating mutation in the DNMT3A gene, which functions as a tumor suppressor and DNA methyltransferase. Functional evidence indicates that truncating mutations in DNMT3A are likely to result in nonsense-mediated decay and haploinsufficiency. This leads to disturbed methylation patterns and global hypomethylation, which are implicated in the pathogenesis of acute myeloid leukemia and other hematologic malignancies. Therefore, the DNMT3A P700Hfs*5 variant is likely to have a damaging effect.
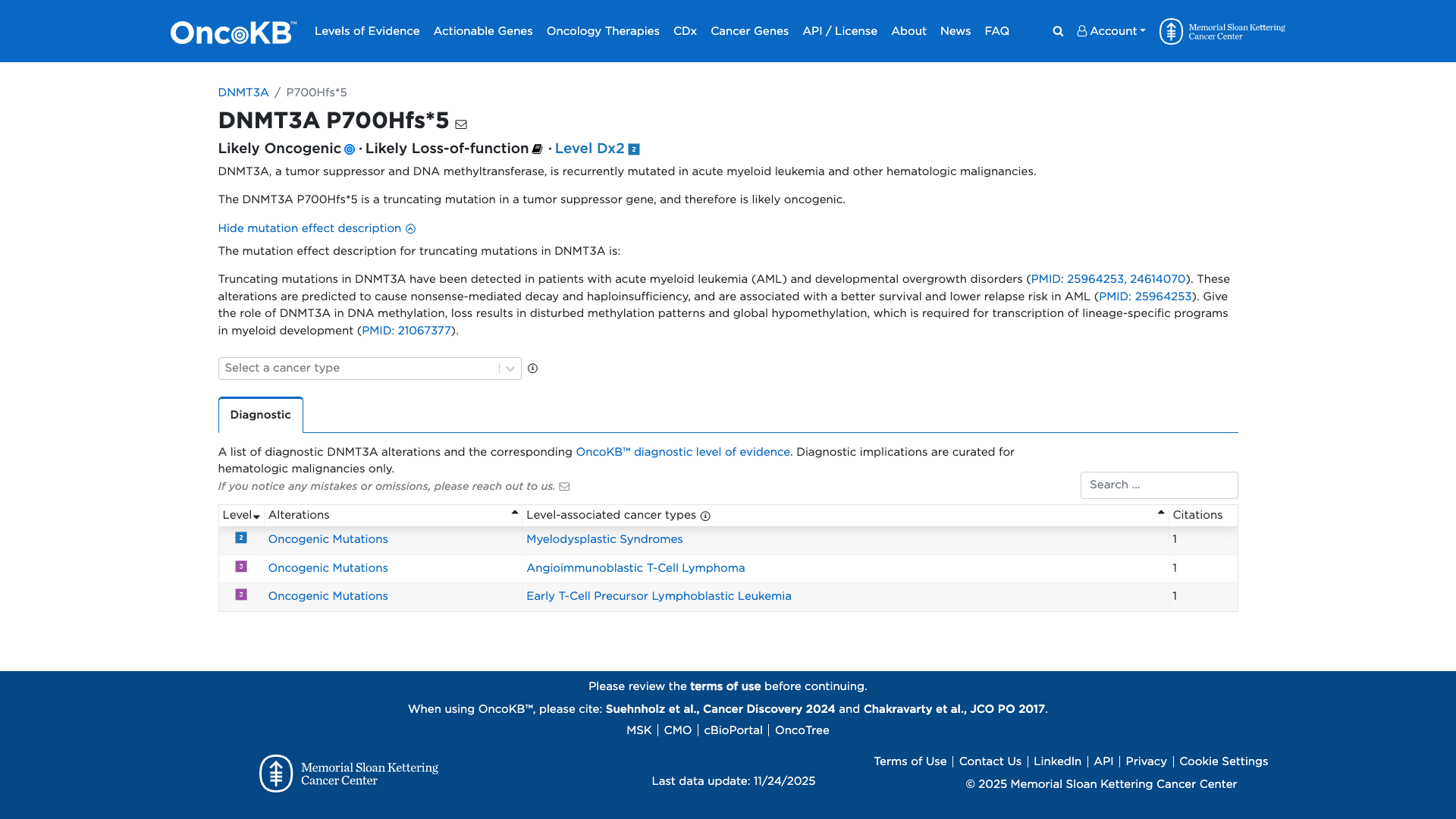
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | 396 bp |
| Donor Loss (DL) | 0.01 | 308 bp |
| Acceptor Gain (AG) | 0.09 | 48 bp |
| Donor Gain (DG) | 0.0 | -73 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to standard ACMG guidelines, the rule for PVS1 is: "Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single exon or multiexon deletion) in a gene where loss of function is a known mechanism of disease." The evidence for this variant shows: c.2099delC causes a frameshift leading to P700Hfs*5, predicted to result in nonsense-mediated decay in DNMT3A, a gene with established LOF mechanism. Therefore, this criterion is applied at Very Strong strength because the variant is a null frameshift in a LOF gene.
PS1 (Not Applied)
According to standard ACMG guidelines, the rule for PS1 is: "Same amino acid change as a known pathogenic variant but different nucleotide change." The evidence for this variant shows: no other pathogenic variant with the same amino acid change exists. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, the rule for PS2 is: "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history." The evidence for this variant shows: no de novo data available. Therefore, this criterion is not applied.
PS3 (Strong)
According to standard ACMG guidelines, the rule for PS3 is: "Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product." The evidence for this variant shows: functional studies demonstrate that truncating DNMT3A variants lead to nonsense-mediated decay, haploinsufficiency, disturbed methylation and leukemogenesis. Therefore, this criterion is applied at Strong strength because well-established functional data support a damaging effect.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "Prevalence in affected individuals is significantly increased compared with controls." The evidence for this variant shows: no case–control or cohort data demonstrating significant enrichment in affected individuals. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot or well-established functional domain without benign variation." The evidence for this variant shows: no data indicating location in a known hot spot or domain. Therefore, this criterion is not applied.
PM2 (Moderate)
According to standard ACMG guidelines, the rule for PM2 is: "Absent from controls (or at extremely low frequency if recessive)." The evidence for this variant shows: gnomAD MAF = 0.000398% (1/251,442 alleles), absent in homozygous state. Therefore, this criterion is applied at Moderate strength because the variant is absent or extremely rare in control populations.
PM3 (Not Applied)
According to standard ACMG guidelines, the rule for PM3 is: "Detected in trans with a pathogenic variant for recessive disorders." The evidence for this variant shows: DNMT3A-related disease is dominant and no trans data. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame deletions/insertions or stop-loss variants." The evidence for this variant shows: it is a frameshift resulting in premature stop, not an in-frame alteration. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, the rule for PM5 is: "Novel missense change at an amino acid residue where a different pathogenic missense change has been seen." The evidence for this variant shows: it is not a missense change. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo, without confirmation of paternity and maternity." The evidence for this variant shows: no de novo data provided. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, the rule for PP1 is: "Co-segregation with disease in multiple affected family members." The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease." The evidence for this variant shows: it is a frameshift, not a missense. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to standard ACMG guidelines, the rule for PP3 is: "Multiple lines of computational evidence support a deleterious effect on the gene/gene product." The evidence for this variant shows: computational tools (CADD 8.33, SpliceAI 0.09) do not predict deleterious effect. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 is: "Patient's phenotype or family history is highly specific for a disease with a single genetic etiology." The evidence for this variant shows: no phenotype/family data provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as pathogenic, but without accessible evidence." The evidence for this variant shows: not reported in ClinVar or other. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to standard ACMG guidelines, the rule for BA1 is: "Allele frequency is too high for the disorder." The evidence for this variant shows: MAF <0.1%, below BA1 threshold. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to standard ACMG guidelines, the rule for BS1 is: "Allele frequency is greater than expected for the disorder." The evidence for this variant shows: allele frequency is very low. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 is: "Observed in healthy individuals with full penetrance expected at an early age." The evidence for this variant shows: no data in healthy subjects. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to standard ACMG guidelines, the rule for BS3 is: "Well-established functional studies show no damaging effect on protein function or splicing." The evidence for this variant shows: functional studies indicate damaging effect. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: "Lack of segregation in affected family members." The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where only LoF causes disease." The evidence for this variant shows: it is a frameshift. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for dominant disorders or in cis with a pathogenic variant." The evidence for this variant shows: no data on phase with other variants. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without known function." The evidence for this variant shows: it is a frameshift, not in-frame. Therefore, this criterion is not applied.
BP4 (Supporting)
According to standard ACMG guidelines, the rule for BP4 is: "Multiple lines of computational evidence suggest no impact." The evidence for this variant shows: CADD score 8.33 (below pathogenic threshold), SpliceAI 0.09 (minimal splicing impact). Therefore, this criterion is applied at Supporting strength because computational predictions do not support a deleterious effect.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease." The evidence for this variant shows: no such case information. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign, but without accessible evidence." The evidence for this variant shows: not reported as benign. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 is: "Synonymous variant with no predicted impact on splicing." The evidence for this variant shows: it is not synonymous. Therefore, this criterion is not applied.