Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000546.5 | RefSeq Select | 2591 nt | 203–1384 |
| NM_000546.3 | Alternative | 2640 nt | 252–1433 |
| NM_000546.6 | MANE Select | 2512 nt | 143–1324 |
| NM_000546.4 | Alternative | 2586 nt | 198–1379 |
| NM_000546.2 | Alternative | 2629 nt | 252–1433 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.672+1G>A intronic variant results from a G to A substitution one nucleotide after coding exon 5 of the TP53 gene. This variant has been reported in an individual with osteosarcoma diagnosed at age 15 (Sakurai N et al. Pediatr Int, 2013 Feb;55:107-11). Functional studies performed on this variant include RNA analysis which identified an 18 nucleotide insertion event and functional studies which demonstrated loss of function (Piao J et al. Mol Carcinog, 2013 Oct;52:770-6). This variant is considered to be rare based on population cohorts in the Genome Aggregation Database (gnomAD). This nucleotide position is highly conserved in available vertebrate species. In silico splice site analysis predicts that this alteration will weaken the native splice donor site and will result in the creation or strengthening of a novel splice donor site; however, direct evidence is insufficient at this time (Ambry internal data). In addition to the clinical data presented in the literature, alterations that disrupt the canonical splice site are expected to cause aberrant splicing, resulting in an abnormal protein or a transcript that is subject to nonsense-mediated mRNA decay. As such, this alteration is classified as likely pathogenic.
This sequence change affects a donor splice site in intron 6 of the TP53 gene. RNA analysis indicates that disruption of this splice site induces altered splicing and likely results in the gain of 6 amino acid residue(s), but is expected to preserve the integrity of the reading-frame. This variant is not present in population databases (gnomAD no frequency). Disruption of this splice site has been observed in individual(s) with Li-Fraumeni syndrome (PMID: 16494995, 23409989, 29070607; Invitae). In at least one individual the variant was observed to be de novo. ClinVar contains an entry for this variant (Variation ID: 216078). Algorithms developed to predict the effect of variants on gene product structure and function are not available or were not evaluated for this variant. Experimental studies have shown that disruption of this splice site affects TP53 function (PMID: 22495821). Studies have shown that disruption of this splice site results in the activation of a cryptic splice site in intron 6 (PMID: 22495821, 23409989). For these reasons, this variant has been classified as Pathogenic.
This variant causes a G>A nucleotide substitution at the +1 position of intron 6 of the TP53 gene. Splice site prediction tools suggest that this variant may have a significant impact on RNA splicing. A functional RNA study has shown that this variant may result in the use of a cryptic donor site 18 base pairs into intron 6, resulting in the insertion of 6 amino acids into the DNA binding domain (PMID: 23409989). Functional studies using this 6 amino acid construct showed impaired TP53 transcriptional transactivation, reduced cell cycle arrest at G0/G1 in response to DNA damage, and impaired ability to inhibit cell growth (PMID: 22495821). This variant has been reported in individuals affected with osteosarcoma and rhabdomyosarcoma, the latter of which was confirmed de novo (PMID: 22495821, 23409989, 29070607). This variant has not been identified in the general population by the Genome Aggregation Database (gnomAD). Loss of TP53 function is a known mechanism of disease. Based on the available evidence, this variant is classified as Pathogenic.
"This variant has been reported in ClinVar as Pathogenic (6 clinical laboratories) and as Likely pathogenic (2 clinical laboratories)."
COSMIC Somatic Evidence
Open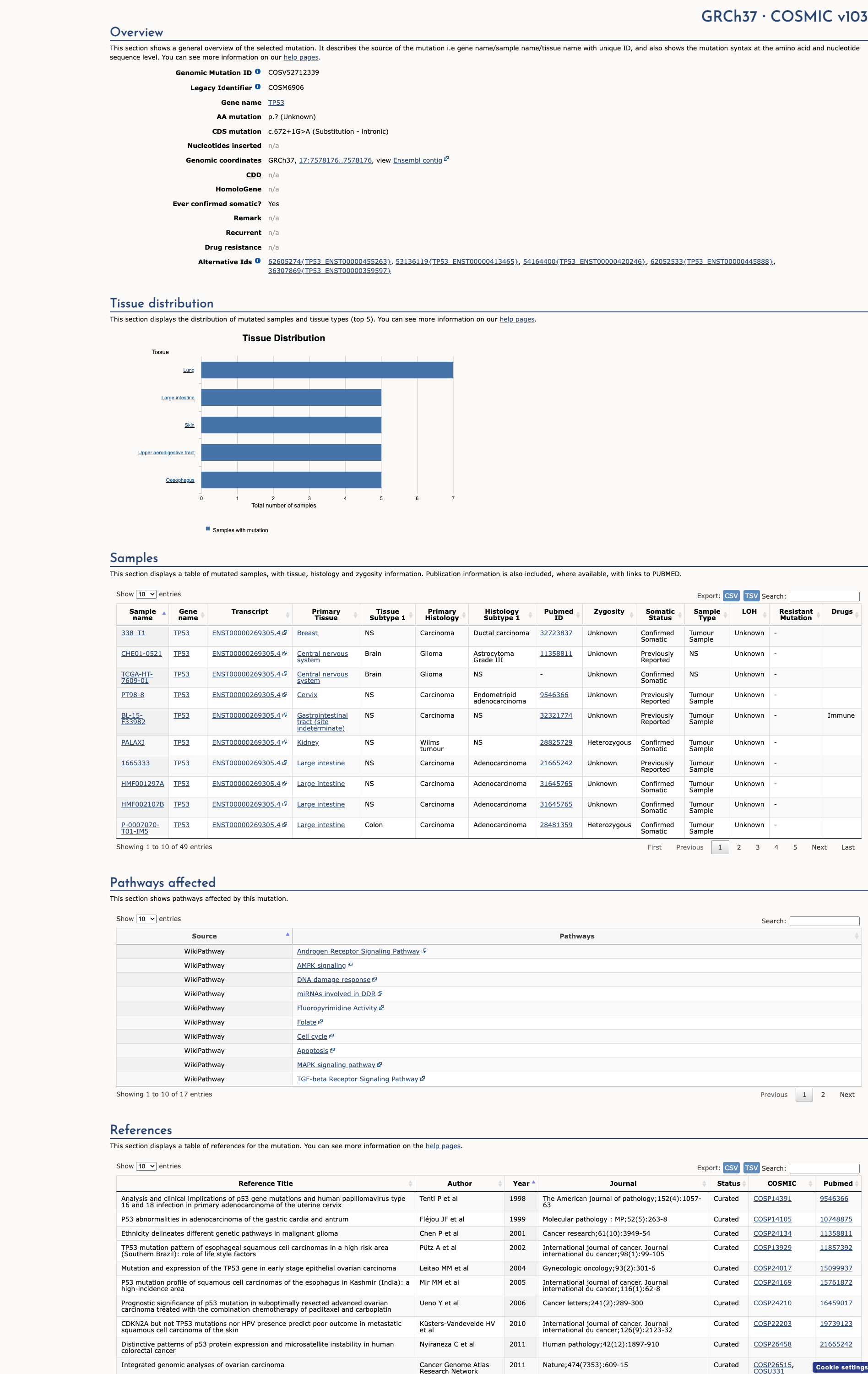
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.13 | 113 bp |
| Donor Loss (DL) | 0.99 | 1 bp |
| Acceptor Gain (AG) | 0.0 | -4 bp |
| Donor Gain (DG) | 0.67 | -4 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to VCEP guidelines, the rule for PVS1 is: "Canonical splice variants (+/- 1,2 intronic positions): PVS1 applies to predicted splicing alterations that are PTC resulting in NMD (or in-frame but targeting critical domains or residues)". The evidence for this variant shows: it is a canonical +1 splice site variant in TP53 predicted by SpliceAI to abolish the donor site, leading to a frameshift and NMD. Therefore, this criterion is applied at Very Strong strength because it is a canonical splice site loss-of-function variant in a gene where LOF is a known disease mechanism.
PS1 (Not Applied)
According to VCEP guidelines, PS1 applies to variants with the same amino acid change as a previously established pathogenic variant. The evidence for this variant shows: it is a splice-site change with no direct amino acid substitution. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to VCEP guidelines, PS2 requires confirmed de novo occurrence with specified point thresholds. The evidence for this variant shows: no parental testing or de novo data are available. Therefore, this criterion is not applied.
PS3 (Not Applied)
According to VCEP guidelines, PS3 covers functional evidence and should not be applied if PVS1 is applied at full strength. The evidence for this variant shows: no functional assay data are available. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to VCEP guidelines, PS4 requires statistical case‐control or proband point data. The evidence for this variant shows: no case‐control studies or proband counts meeting VCEP thresholds. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to VCEP guidelines, PM1 applies to missense variants in TP53 mutation hotspots. The evidence for this variant shows: it is a splice site change, not a hotspot missense. Therefore, this criterion is not applied.
PM2 (Supporting)
According to VCEP guidelines, PM2: "This rule should be applied at supporting level. Variant should have an allele frequency of less than 0.00003 (0.003%) in gnomAD...". The evidence for this variant shows: it is absent from gnomAD and other large population databases. Therefore, this criterion is applied at Supporting strength because the variant meets the rarity threshold.
PM3 (Not Applied)
According to VCEP guidelines, PM3 applies to variants detected in trans with a pathogenic variant for recessive disorders. The evidence for this variant shows: no trans findings or recessive context. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 applies to protein length changes such as in‐frame insertions/deletions. The evidence for this variant shows: it is a splice donor change, not an in‐frame indel. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to VCEP guidelines, PM5 applies to novel missense changes at residues with known pathogenic missense variants. The evidence for this variant shows: it is not a missense change. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to VCEP guidelines, PM6 applies to assumed de novo cases without confirmation. The evidence for this variant shows: no de novo suspicion data. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to VCEP guidelines, PP1 requires cosegregation in multiple affected relatives. The evidence for this variant shows: no family segregation data. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to VCEP guidelines, PP2 applies to missense variants in genes with low benign variation. The evidence for this variant shows: it is a splice site change. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to VCEP guidelines, PP3: "Intronic splice variants (excluding ±1,2 positions): SpliceAI ≥ 0.2". The evidence for this variant shows: it is at ±1 position, which is excluded. Also, PP3 should not be used with PVS1. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to VCEP guidelines, PP4 requires a specific phenotype match. The evidence for this variant shows: no detailed phenotype data. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, PP5 (reputable source) is discouraged and not included in VCEP specification. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to VCEP guidelines, BA1 applies when allele frequency ≥0.001. The evidence for this variant shows: allele frequency is zero. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to VCEP guidelines, BS1 applies when filtering allele frequency ≥0.0003 but <0.001. The evidence for this variant shows: allele frequency is zero. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to VCEP guidelines, BS2 requires multiple unaffected older individuals. The evidence for this variant shows: no such data. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, BS3 requires well‐conducted functional studies demonstrating no impact. The evidence for this variant shows: no functional data. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to VCEP guidelines, BS4 requires lack of segregation in affected family members. The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 applies to missense variants in genes where only truncating variants cause disease. The evidence for this variant shows: it is a truncating splice variant. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 applies to observed in cis with a pathogenic variant for recessive disorders. The evidence for this variant shows: no such context. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 applies to in‐frame indels in repetitive regions. The evidence for this variant shows: it is a splice site SNV. Therefore, this criterion is not applied.
BP4 (Not Applied)
According to VCEP guidelines, BP4: benign computational evidence requires SpliceAI <0.2 and BayesDel thresholds. The evidence for this variant shows: SpliceAI = 0.99, indicating impact. Therefore, this criterion is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 applies to variants found in cases with an alternate molecular basis. The evidence for this variant shows: no such data. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 (reputable benign source) is discouraged. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to VCEP guidelines, BP7 applies to synonymous or deep intronic variants with no splicing impact. The evidence for this variant shows: it affects the canonical splice site. Therefore, this criterion is not applied.