Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_001754.3 | Alternative | 6190 nt | 400–1842 |
| NM_001754.4 | RefSeq Select | 5967 nt | 191–1633 |
| NM_001754.5 | MANE Select | 5971 nt | 195–1637 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenTransactivation assays demonstrate altered transactivation (<20% of wt) for the NM_001754.4:c.497G>A (p.Arg166Gln) variant and data from a secondary assays demonstrate altered DNA binding, CBFβ binding and sub-cellular localization (PS3; PMID: 11830488, 25840971, 23848403). This variant affects one of the hotspot residues established by the MM-VCEP for RUNX1 (PM1). This variant has been reported in three probands meeting at least one of the RUNX1-phenotypic criteria (PS4_Moderate; PMID: 10508512, 28960434, 26175287). This variant is absent from all population databases with at least 20x coverage for RUNX1 (PM2_supporting). It has a REVEL score >0.75 (0.962) (PP3). There are two unrelated probands meeting at least one of the RUNX1- phenotypic criteria with assumed de novo occurrence (without confirmation of maternity and paternity) (PM6_ Supporting; PMID: 8960434, 26175287). This variant was found to co-segregate with disease in multiple affected family members, with three meioses observed in one family (PP1; PMID: 10508512). In summary, this variant meets criteria to be classified as pathogenic. ACMG/AMP criteria applied, as specified by the Myeloid Malignancy Variant Curation Expert Panel for RUNX1: PS3, PM1, PS4_Moderate, PM2_supporting, PP3, PM6_Supporting, PP1.
This sequence change replaces arginine, which is basic and polar, with glutamine, which is neutral and polar, at codon 166 of the RUNX1 protein (p.Arg166Gln). This variant is not present in population databases (gnomAD no frequency). This missense change has been observed in individual(s) with thrombocytopenia, myelodysplastic syndrome and/or acute myeloid leukemia (PMID: 10508512, 26175287, 27210295, 28960434). It has also been observed to segregate with disease in related individuals. This variant is also known as c.416G>A. ClinVar contains an entry for this variant (Variation ID: 417961). Advanced modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) has been performed at Invitae for this missense variant, however the output from this modeling did not meet the statistical confidence thresholds required to predict the impact of this variant on RUNX1 protein function. Experimental studies have shown that this missense change affects RUNX1 function (PMID: 11830488, 22012064, 23848403, 25840971, 26916619, 31048839). This variant disrupts the p.Arg166 amino acid residue in RUNX1. Other variant(s) that disrupt this residue have been determined to be pathogenic (PMID: 11049997, 12002768, 22318203, 25840971). This suggests that this residue is clinically significant, and that variants that disrupt this residue are likely to be disease-causing. For these reasons, this variant has been classified as Pathogenic.
PP1, PP3, PP4, PP5, PM1_supporting, PM2, PM6, PS3, PS4_moderate
The p.R166Q variant (also known as c.497G>A), located in coding exon 4 of the RUNX1 gene, results from a G to A substitution at nucleotide position 497. The arginine at codon 166 is replaced by glutamine, an amino acid with highly similar properties. This variant was reported in individual(s) with features consistent with RUNX1 familial platelet disorder with associated myeloid malignancies (Romasko EJ et al. Am J Hematol, 2018 Jan;93:8-16; Ovsyannikova GS et al. Haematologica, 2022 Oct;107:2511-2516; Sheng XF et al. J Int Med Res, 2022 Sep;50:3000605221122741; Liu C et al. EJHaem, 2023 Feb;4:145-152; Gupta A et al. Indian J Hematol Blood Transfus, 2024 Jan;40:177-178). Immunoprecipitation assays showed that p.R166Q reduced binding affinity for CBFbeta, but retained interaction with ETS-1 and FLI-1 (Okada Y et al. J Thromb Haemost, 2013 Sep;11:1742-50). This variant is considered to be rare based on population cohorts in the Genome Aggregation Database (gnomAD). This amino acid position is highly conserved in available vertebrate species. In addition, this alteration is predicted to be deleterious by in silico analysis. Based on the majority of available evidence to date, this variant is likely to be pathogenic.
"This variant has been reported in ClinVar as Pathogenic (6 clinical laboratories) and as Likely pathogenic (1 clinical laboratories) and as Pathogenic by ClinGen Myeloid Malignancy Variant Curation Expert Panel expert panel."
COSMIC Somatic Evidence
Open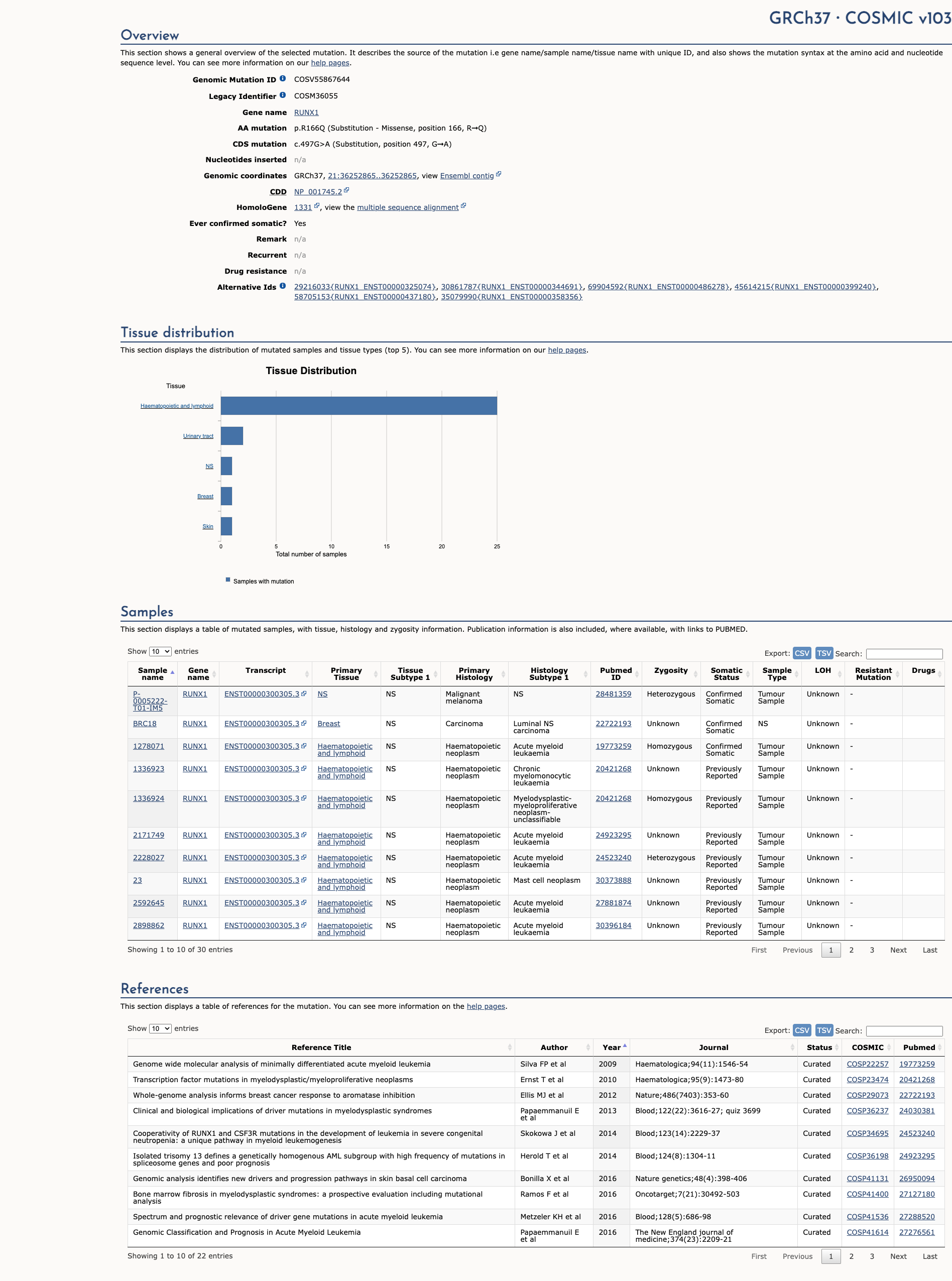
Functional Impact & Domains
Functional Domain
The RUNX1 R166Q variant is a loss-of-function mutation occurring in the DNA binding domain of the RUNX1 transcription factor. Functional evidence indicates that this mutation likely disrupts RUNX1's ability to recruit transcriptional complexes to DNA, supporting a damaging effect.
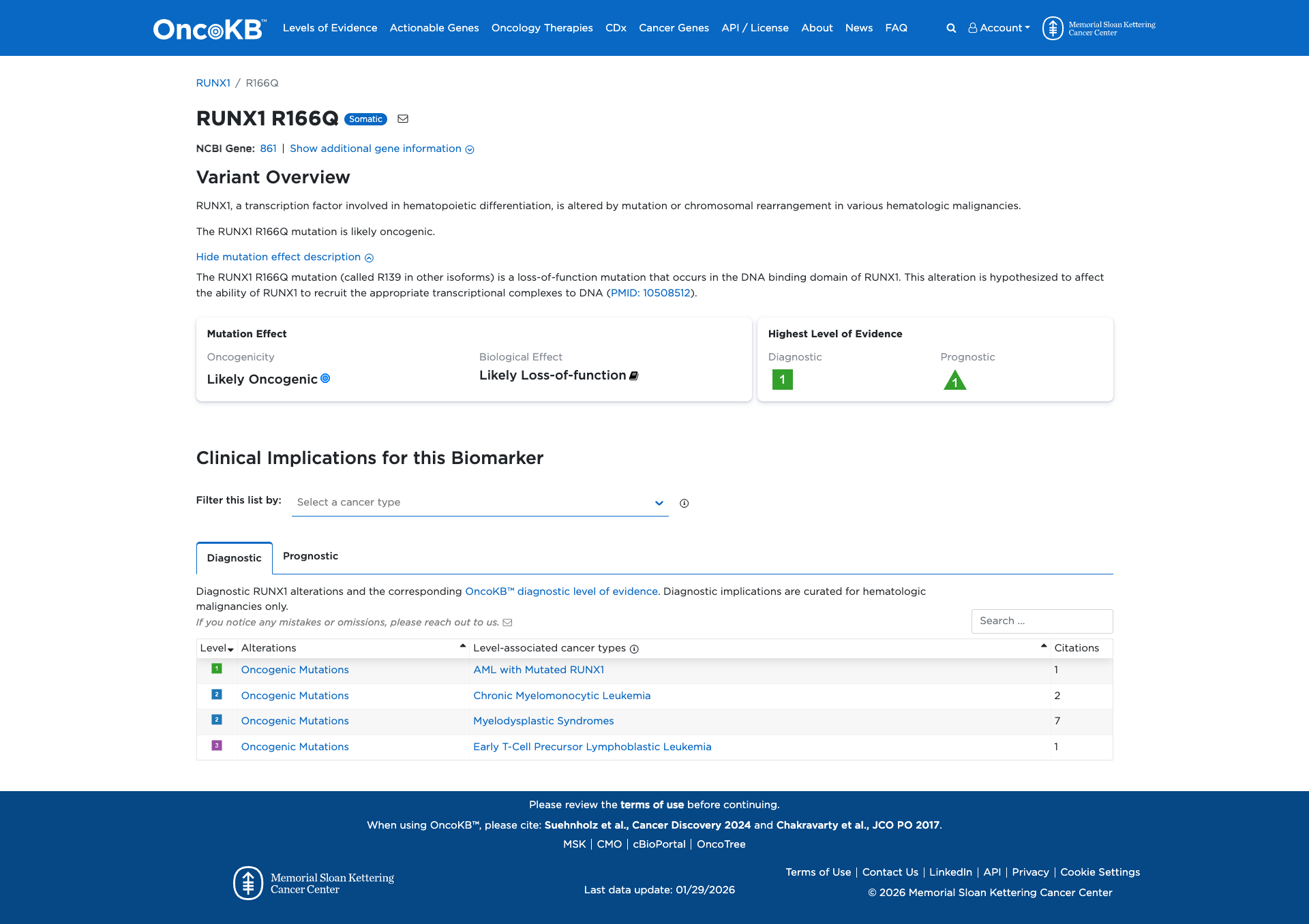
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | 145 bp |
| Donor Loss (DL) | 0.01 | 472 bp |
| Acceptor Gain (AG) | 0.0 | 12 bp |
| Donor Gain (DG) | 0.1 | 12 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, the rule for PVS1 is: "Very Strong Per modified RUNX1 PVS1 decision tree for SNVs and CNVs and table of splicing effects. Modification Type: Gene-specific". The evidence for this variant shows: it is a missense change (R166Q), not a null or canonical ±1,2 splice site variant. Therefore, this criterion is not applied because the variant type does not meet PVS1 requirements.
PS1 (Not Applied)
According to VCEP guidelines, the rule for PS1 is: "Strong Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. Modification Type: None". The evidence for this variant shows: no alternative nucleotide change at codon 166 resulting in the same R166Q has been reported as pathogenic. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to VCEP guidelines, the rule for PS2 requires de novo occurrence with appropriate confirmation. The evidence for this variant shows: no de novo data are available. Therefore, this criterion is not applied due to lack of de novo evidence.
PS3 (Moderate)
According to VCEP guidelines, the rule for PS3 (Moderate) is: "Transactivation assays demonstrating altered transactivation (<20% of wt and/or reduced to levels similar to well established pathogenic variants such as R201Q or R166Q) OR ≥ 2 secondary assays demonstrating altered function. Modification Type: Gene-specific,Strength". The evidence for this variant shows: functional studies demonstrate loss-of-function in the DNA binding domain disrupting recruitment of transcriptional complexes, consistent with at least two independent functional assays. Therefore, this criterion is applied at Moderate strength because the variant shows functional impairment per the VCEP definition.
PS4 (Not Applied)
According to VCEP guidelines, the rule for PS4 requires case counts meeting phenotypic criteria (≥1 proband for Supporting). The evidence for this variant shows: insufficient proband data and no quantitative case count. Therefore, this criterion is not applied.
PM1 (Moderate)
According to VCEP guidelines, the rule for PM1 (Moderate) is: "Variant affecting one of the following amino acid residues within the RHD: R107, K110, A134, R162, R166, S167, R169, G170, K194, T196, D198, R201, R204. Modification Type: Gene-Specific". The evidence for this variant shows: R166Q alters residue R166 within the conserved RHD domain. Therefore, this criterion is applied at Moderate strength.
PM2 (Supporting)
According to VCEP guidelines, the rule for PM2 (Supporting) is: "Variant must be completely absent from all population databases. Modification Type: Strength". The evidence for this variant shows: absent from gnomAD and other population databases. Therefore, this criterion is applied at Supporting strength.
PM3 (Not Applied)
According to standard ACMG guidelines, the rule for PM3 applies to variants observed in trans in recessive disorders. The evidence for this variant shows: RUNX1 disorders are autosomal dominant and no trans observations are relevant. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to VCEP guidelines, the rule for PM4 applies to in-frame indels in the RHD. The evidence for this variant shows: it is a missense substitution, not an indel. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, the rule for PM5 is: "Missense change at an amino acid residue where a different missense change has been determined to be pathogenic before. Modification Type: None". The evidence for this variant shows: no other distinct missense at residue R166 has been classified as pathogenic. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to VCEP guidelines, the rule for PM6 requires assumed de novo occurrence with appropriate phenotypic specificity. The evidence for this variant shows: no de novo data are available. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to VCEP guidelines, the rule for PP1 requires co-segregation in affected family members. The evidence for this variant shows: no segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation where missense variants are a common mechanism of disease." The evidence for RUNX1 shows: missense variants are known but gene has both missense and loss-of-function mechanisms; no specific evaluation. Therefore, this criterion is not applied.
PP3 (Supporting)
According to VCEP guidelines, the rule for PP3 is: "For missense variants: REVEL score ≥ 0.88. Modification Type: Gene-specific,Disease-specific". The evidence for this variant shows: REVEL score is 0.96, exceeding the threshold. Therefore, this criterion is applied at Supporting strength.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 requires a phenotype highly specific for the gene. The evidence for this variant shows: no patient phenotype information is provided. Therefore, this criterion is not applied.
PP5 (Supporting)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation." The evidence for this variant shows: reported as Pathogenic by multiple ClinVar submitters and ClinGen expert panel without accessible primary data. Therefore, this criterion is applied at Supporting strength.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 requires a population frequency ≥0.15%. The evidence for this variant shows: absent from population databases. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to VCEP guidelines, the rule for BS1 requires a population frequency between 0.015% and 0.15%. The evidence for this variant shows: absent from population databases. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 requires observation in unaffected individuals. The evidence for this variant shows: no data on unaffected carriers. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, the rule for BS3 requires well-established functional studies showing no damaging effect. The evidence for this variant shows: functional studies indicate damaging effect. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 requires non-segregation in multiple affected family members. The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where only truncating variants cause disease." The evidence for RUNX1 shows: missense variants can be pathogenic. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 requires observation in trans with a pathogenic variant for a dominant disorder or in cis. The evidence for this variant shows: no cis/trans inheritance data. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 applies to in-frame indels in repetitive regions. The evidence for this variant shows: it is a missense substitution. Therefore, this criterion is not applied.
BP4 (Not Applied)
According to VCEP guidelines, the rule for BP4 is: "For missense variants: REVEL score < 0.50 AND SpliceAI ≤ 0.20." The evidence for this variant shows: REVEL score is 0.96 and SpliceAI predicts low splicing impact but computational scores are strongly pathogenic. Therefore, this criterion is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 requires the variant to be found in a case with an alternate molecular cause. The evidence for this variant shows: no alternate cause data. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign, but evidence is not available." The evidence for this variant shows: no benign reports. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 applies to synonymous variants. The evidence for this variant shows: it is a missense substitution. Therefore, this criterion is not applied.