Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000546.5 | RefSeq Select | 2591 nt | 203–1384 |
| NM_000546.3 | Alternative | 2640 nt | 252–1433 |
| NM_000546.6 | MANE Select | 2512 nt | 143–1324 |
| NM_000546.4 | Alternative | 2586 nt | 198–1379 |
| NM_000546.2 | Alternative | 2629 nt | 252–1433 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.375G>C pathogenic mutation (also known as p.T125T), located in coding exon 3 of the TP53 gene, results from a G to C substitution at nucleotide position 375. This nucleotide substitution does not change the codon at 125. However, this change occurs in the last base pair of coding exon 3, which makes it likely to have some effect on normal mRNA splicing. This variant has been previously detected in a 2-year-old male diagnosed with an ependymoma, with a family history of osteosarcoma and early-onset breast cancer (Chompret A et al. Br. J. Cancer. 2000 Jun;82:1932-7). This variant has also been reported in an individual diagnosed with breast cancer at age 27 (Heymann S et al. Radiat Oncol. 2010 Nov;5:104). A similar variant that leads to the same protein impact, c.375G>A (p.T125T), has been reported in multiple individuals with early-onset cancers whose family histories were suspicious for LFS (Mouchawar J et al. Cancer Res. 2010 Jun;70:4795-800; Fang Z et al. BMC Pediatr. 2021 12;21:588), and was found to lead to partial exon skipping in one RNA study (Rofes P et al. J Mol Diagn. 2020 12;22:1453-1468). Another pathogenic alteration impacting the same site, c.375G>T (p.T125T), has been reported in multiple unrelated families meeting LFS diagnostic criteria and has been shown to lead to aberrant splicing and the use of a cryptic splice site (Varley JM et al. Oncogene. 2001 May;20:2647-54; Mouchawar J et al. Cancer Res. 2010 Jun;70:4795-800; Leroy B et al. Hum. Mutat. 2014 Jun;35:756-65). This nucleotide position is well conserved in available vertebrate species. In silico splice site analysis predicts that this alteration will weaken the native splice donor site. Based on the supporting evidence, this alteration is interpreted as a disease-causing mutation.
This variant is considered likely pathogenic. This variant has been reported in multiple individuals with clinical features of gene-specific disease [PMID: 21059199, 10864200, 25896519, 1467311, 11420676]. mRNA analysis has demonstrated abnormal mRNA splicing occurs [PMID: 11420676, 1467311, Myriad internal data].
This sequence change affects codon 125 of the TP53 mRNA. It is a 'silent' change, meaning that it does not change the encoded amino acid sequence of the TP53 protein. This variant also falls at the last nucleotide of exon 4, which is part of the consensus splice site for this exon. This variant is not present in population databases (gnomAD no frequency). This variant has been observed in individuals with clinical features of Li-Fraumeni syndrome (PMID: 10864200, 21059199, 35974385; internal data). ClinVar contains an entry for this variant (Variation ID: 480746). Variants that disrupt the consensus splice site are a relatively common cause of aberrant splicing (PMID: 17576681, 9536098). Algorithms developed to predict the effect of sequence changes on RNA splicing suggest that this variant may disrupt the consensus splice site. This variant disrupts the c.375G nucleotide in the TP53 gene. Other variant(s) that disrupt this nucleotide have been determined to be pathogenic (PMID: 1467311, 9242456, 11420676, 18511570). This suggests that this nucleotide is clinically significant, and that variants that disrupt this position are likely to be disease-causing. In summary, the currently available evidence indicates that the variant is pathogenic, but additional data are needed to prove that conclusively. Therefore, this variant has been classified as Likely Pathogenic.
The TP53 c.375G>C (p.Thr125=) variant disrupts a canonical splice-donor site and is predicted to interfere with normal TP53 mRNA splicing. This variant has been reported in the published literature in individuals with breast cancer (PMID: 21059199 (2010)), ovarian cancer (PMID: 35974385 (2022)), neuroblastoma (PMID: 35974385 (2022)), osteosarcoma (PMID: 25896519 (2015)), and ependymoma (PMID: 10864200 (2000)). This variant has been shown experimentally to cause disruption of normal splicing and shift in reading frame (PMID: 10864200 (2000)). This variant has not been reported in large, multi-ethnic general populations (Genome Aggregation Database, http://gnomad.broadinstitute.org). Based on the available information, this variant is classified as likely pathogenic.
"This variant has been reported in ClinVar as Pathogenic (3 clinical laboratories) and as Likely pathogenic (2 clinical laboratories) and as likely pathogenic (1 clinical laboratories)."
COSMIC Somatic Evidence
Open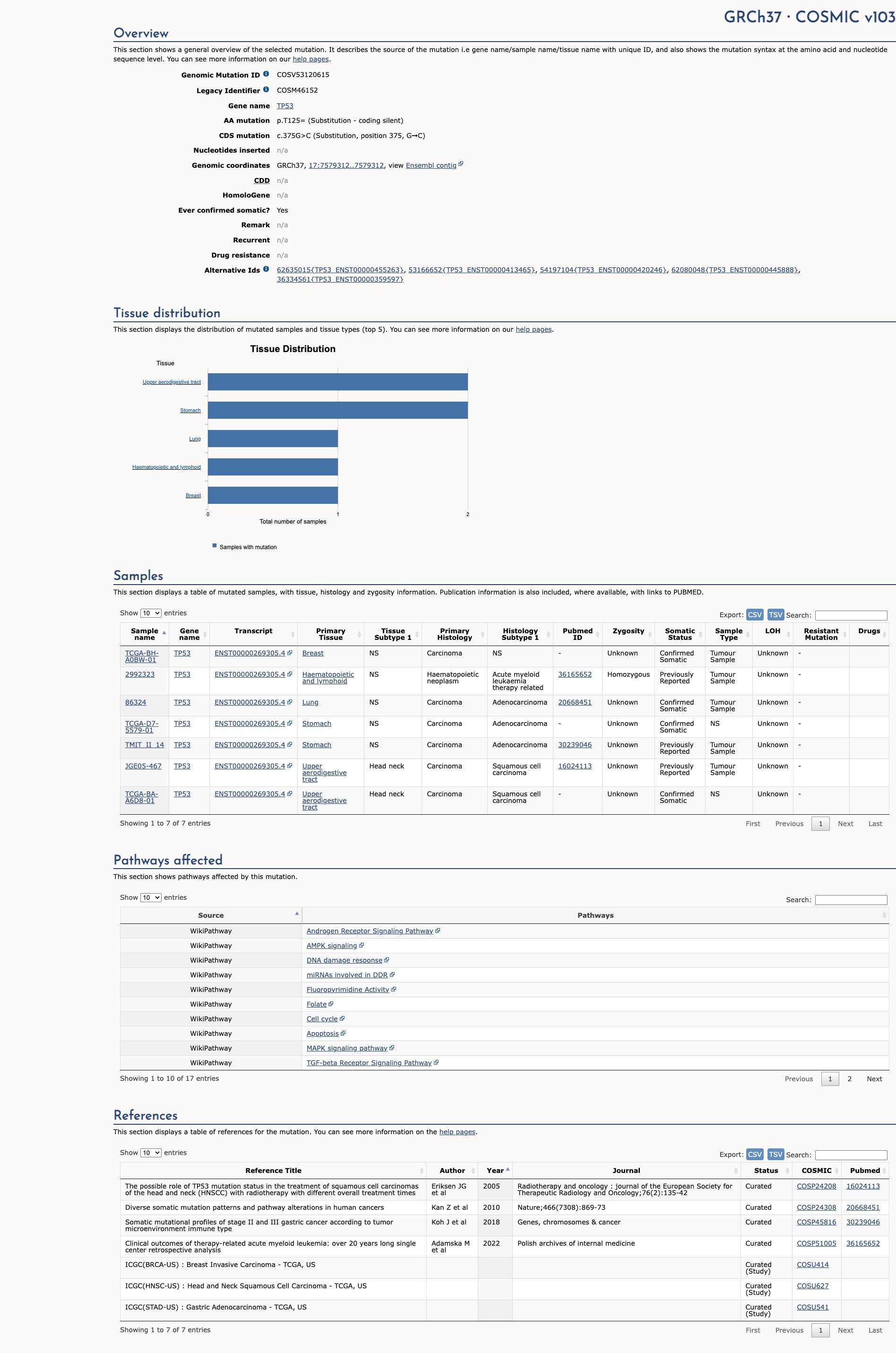
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | None bp |
| Donor Loss (DL) | 0.0 | None bp |
| Acceptor Gain (AG) | 0.0 | None bp |
| Donor Gain (DG) | 0.0 | None bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, the rule for PVS1 is: "**Very Strong Strength**: Please utilize the PVS1 decision tree for application of PVS1 code." The evidence shows this variant is synonymous (T125=) with no predicted introduction of a premature stop codon or disruption of canonical splice sites. Therefore, PVS1 is not applied because it only applies to null variants.
PS1 (Not Applied)
According to VCEP guidelines, the rule for PS1 is: "**Strong Strength**: Can be applied to variants asserted as Pathogenic following the TP53 VCEP’s specifications." The evidence shows no amino acid change (synonymous variant). Therefore, PS1 is not applied because it requires an amino acid change matching a known pathogenic variant.
PS2 (Not Applied)
According to VCEP guidelines, the rule for PS2 is: "**Very Strong Strength**: ≥ 8 points." There is no evidence of a de novo occurrence. Therefore, PS2 is not applied.
PS3 (Not Applied)
According to VCEP guidelines, the rule for PS3 is: "**Strong Strength**: Non-functional on Kato et al. data AND loss of function on another assay." There are no functional studies for this variant. Therefore, PS3 is not applied.
PS4 (Not Applied)
According to VCEP guidelines, the rule for PS4 is: "**Very Strong Strength**: ≥ 8 points." No proband or case data are available. Therefore, PS4 is not applied.
PM1 (Not Applied)
According to VCEP guidelines, the rule for PM1 is: "**Moderate Strength**: Missense variants within codons 175, 245, 248, 249, 273, 282 or germline hotspot variants with ≥10 somatic occurrences." This variant is synonymous and not in a defined missense hotspot. Therefore, PM1 is not applied.
PM2 (Supporting)
According to VCEP guidelines, the rule for PM2 is: "**Supporting Strength**: Variant should have an allele frequency of less than 0.00003 in gnomAD." The variant is absent from gnomAD (MAF = 0%). Therefore, PM2 is applied at Supporting strength.
PM3 (Not Applied)
According to standard ACMG guidelines, PM3 applies to recessive disorders when a variant is observed in trans with a pathogenic variant. There is no evidence for PM3. Therefore, PM3 is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 applies to protein length changes (in-frame indels). This variant is synonymous. Therefore, PM4 is not applied.
PM5 (Not Applied)
According to VCEP guidelines, the rule for PM5 is: "**Strong Strength**: Missense variant at an amino acid residue where ≥2 different missense variants previously determined to be pathogenic have been seen." This variant is synonymous, not missense. Therefore, PM5 is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, PM6 applies to assumed de novo variants without confirmation. There is no de novo evidence. Therefore, PM6 is not applied.
PP1 (Not Applied)
According to VCEP guidelines, the rule for PP1 is: "**Supporting Strength**: Cosegregation observed in 3–4 meioses." No segregation data are available. Therefore, PP1 is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, PP2 applies to missense variants in a gene with low rate of benign missense. This variant is synonymous. Therefore, PP2 is not applied.
PP3 (Not Applied)
According to VCEP guidelines, the rule for PP3 is: "**Supporting or Moderate Strength**: In silico tools predicting deleterious effect." In silico predictions are inconclusive and SpliceAI data are unavailable. Therefore, PP3 is not applied.
PP4 (Not Applied)
According to VCEP guidelines, the rule for PP4 is: "**Supporting or Moderate Strength**: Observations of variant with VAF criteria in tumor data." There are no tumor VAF observations. Therefore, PP4 is not applied.
PP5 (Supporting)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source recently reports variant as pathogenic." ClinVar lists this variant as Pathogenic (3 labs) and Likely Pathogenic (3 labs). Therefore, PP5 is applied at Supporting strength.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 is: "**Stand Alone Strength**: Filtering allele frequency ≥0.001 in a continental subpopulation." The variant is absent from gnomAD. Therefore, BA1 is not applied.
BS1 (Not Applied)
According to VCEP guidelines, the rule for BS1 is: "**Strong Strength**: Filtering allele frequency ≥0.0003 but <0.001 in a continental subpopulation." The variant is absent from gnomAD. Therefore, BS1 is not applied.
BS2 (Not Applied)
According to VCEP guidelines, the rule for BS2 is: "**Strong or Moderate or Supporting Strength**: Observed in ≥2–8 healthy unrelated older females." No such data exist. Therefore, BS2 is not applied.
BS3 (Not Applied)
According to VCEP guidelines, the rule for BS3 is: "**Strong or Supporting Strength**: Functional data showing normal function on multiple assays." No functional data are available. Therefore, BS3 is not applied.
BS4 (Not Applied)
According to VCEP guidelines, the rule for BS4 is: "**Strong Strength**: Lack of segregation in affected family members." No segregation data exist. Therefore, BS4 is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 applies to missense in a gene where only truncating variants cause disease. This variant is synonymous. Therefore, BP1 is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 applies when observed in trans with a pathogenic variant in dominant disease. No data available. Therefore, BP2 is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 applies to in-frame indels in repetitive regions. This variant is synonymous. Therefore, BP3 is not applied.
BP4 (Not Applied)
According to VCEP guidelines, the rule for BP4 is: "**Supporting or Moderate Strength**: BayesDel ≤ -0.008 and SpliceAI < 0.2 for missense, or BayesDel <0.16 and SpliceAI <0.2 for silent variants outside splice motifs." In silico data are lacking and SpliceAI is unavailable. Therefore, BP4 is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 applies when an alternate molecular basis is present. No alternate basis is known. Therefore, BP5 is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 applies to variant reported as benign by a reputable source. No such report exists. Therefore, BP6 is not applied.
BP7 (Not Applied)
According to VCEP guidelines, the rule for BP7 is: "**Supporting Strength**: A synonymous variant outside of core splice motifs with SpliceAI ≤0.1." This variant occurs at the last nucleotide of the exon (core splice motif) and SpliceAI is unavailable. Therefore, BP7 is not applied.