Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000546.5 | RefSeq Select | 2591 nt | 203–1384 |
| NM_000546.3 | Alternative | 2640 nt | 252–1433 |
| NM_000546.6 | MANE Select | 2512 nt | 143–1324 |
| NM_000546.4 | Alternative | 2586 nt | 198–1379 |
| NM_000546.2 | Alternative | 2629 nt | 252–1433 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThis variant is considered likely pathogenic. Functional studies indicate this variant impacts protein function [PMID: 29979965]. This variant is expected to disrupt protein structure [Myriad internal data].
The p.V172F pathogenic mutation (also known as c.514G>T), located in coding exon 4 of the TP53 gene, results from a G to T substitution at nucleotide position 514. The valine at codon 172 is replaced by phenylalanine, an amino acid with highly similar properties. This variant was reported in individual(s) with features consistent with Li-Fraumeni syndrome (Siddiqui R et al. Fam Cancer, 2005;4:177-81; Ricker CA et al. Cold Spring Harb Mol Case Stud, 2021 02;7:). This alteration is located in the highly conserved domain III of the TP53 DNA binding domain. This alteration was shown to have a loss of transactivation capacity in yeast-based functional studies (Kato S et al. Proc Natl Acad Sci USA. 2003 Jul 8;100(14):8424-9). Studies conducted in human cell lines indicate this alteration is deficient at growth suppression and has a dominant negative effect (Kotler E et al. Mol.Cell. 2018 Jul;71:178-190.e8; Giacomelli AO et al. Nat. Genet. 2018 Oct;50:1381-1387). This variant has been detected in at least one individual at an allele fraction that is suggestive of clonal hematopoiesis, a predictor of TP53 pathogenicity (Ambry internal data; Fortuno C et al. Genet Med. 2022 03;24:673-680). This alteration has also been observed numerous times as a somatic mutation in the cancerhotspots.org database (Chang MT et al. Cancer Discov. 2018 02;8:174-183). This variant is considered to be rare based on population cohorts in the Genome Aggregation Database (gnomAD). This amino acid position is highly conserved in available vertebrate species. In addition, this alteration is predicted to be deleterious by in silico analysis. Based on the supporting evidence, this alteration is interpreted as a disease-causing mutation.
This sequence change replaces valine, which is neutral and non-polar, with phenylalanine, which is neutral and non-polar, at codon 172 of the TP53 protein (p.Val172Phe). This variant is not present in population databases (gnomAD no frequency). This missense change has been observed in individual(s) with clinical features of Li-Fraumeni syndrome (PMID: 15951970, 33436392). ClinVar contains an entry for this variant (Variation ID: 428909). Invitae Evidence Modeling incorporating data from in vitro experimental studies (PMID: 12826609, 29979965, 30224644) indicates that this missense variant is expected to disrupt TP53 function with a positive predictive value of 97.5%. Experimental studies have shown that this missense change affects TP53 function (PMID: 21343334). In summary, the available evidence is currently insufficient to determine the role of this variant in disease. Therefore, it has been classified as a Variant of Uncertain Significance.
"This variant has been reported in ClinVar as Likely pathogenic (1 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Pathogenic (1 clinical laboratories)."
COSMIC Somatic Evidence
Open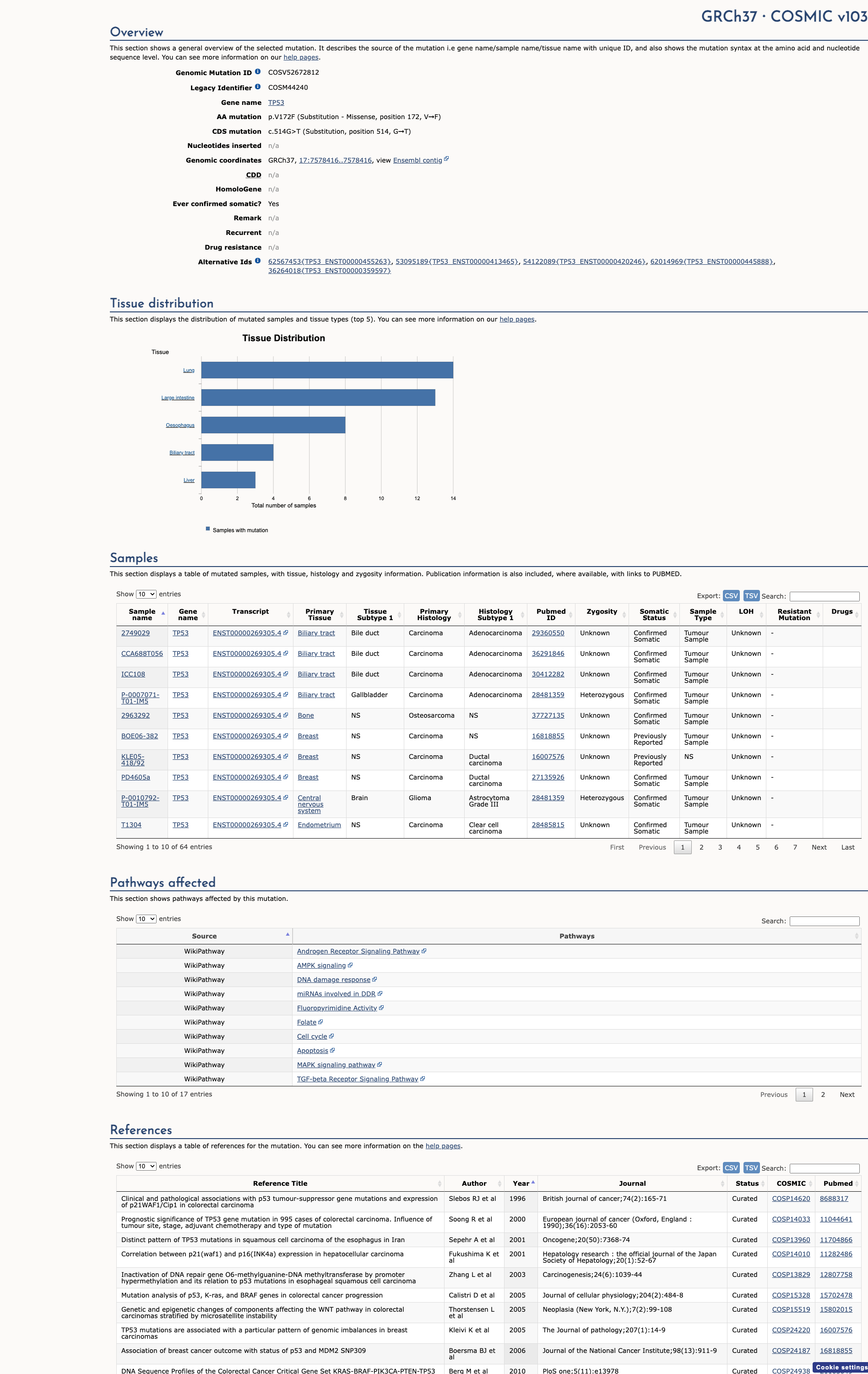
Functional Impact & Domains
Functional Domain
The TP53 V172F variant has been functionally characterized and is shown to be damaging. It is located in the DNA-binding domain of the TP53 protein and results in the loss of transactivational activity, indicating an inactivating effect. Additionally, the variant enhances interaction with Mdm4, leading to context-dependent alterations in TP53 stability, further supporting a loss of TP53 protein function.
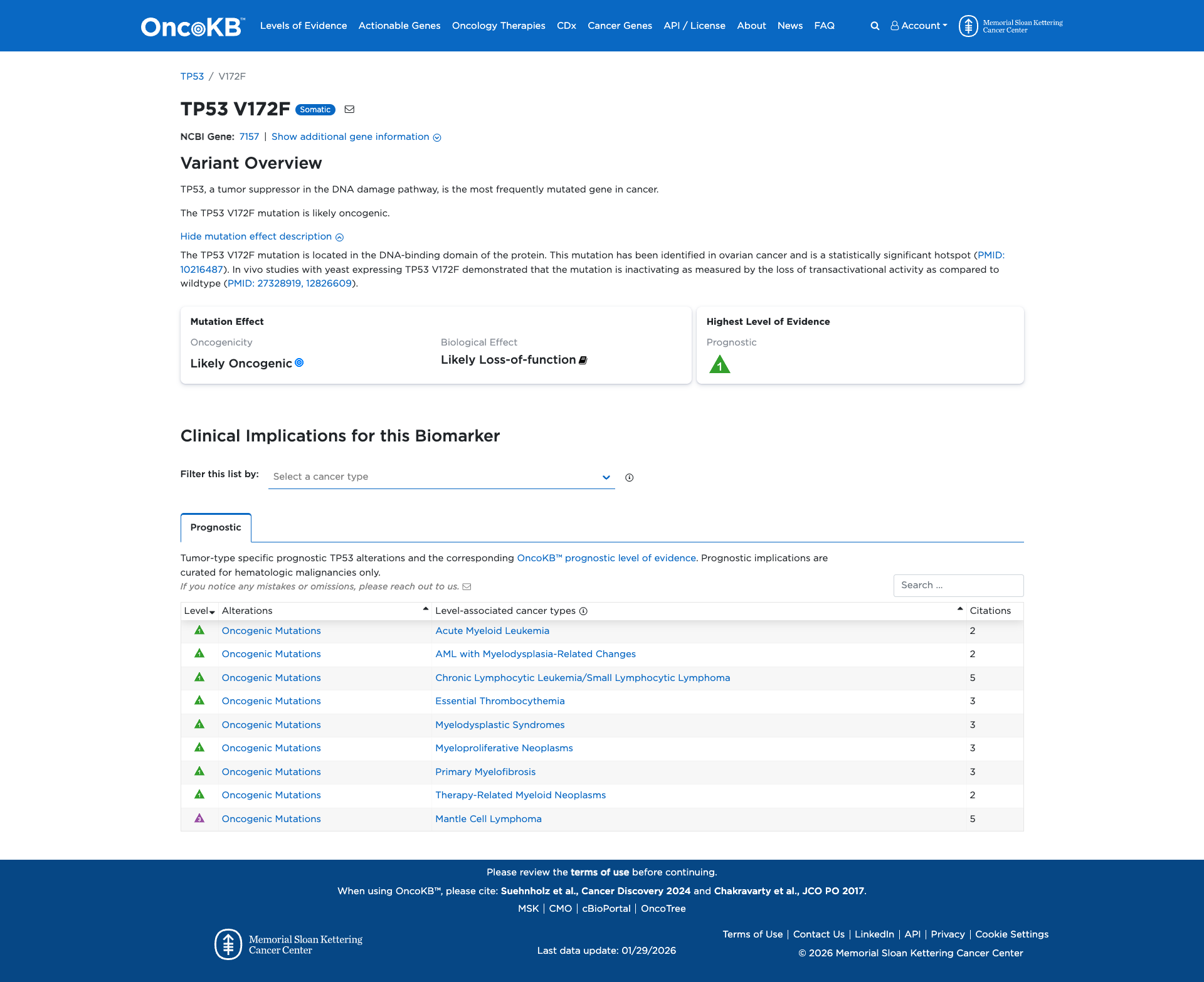
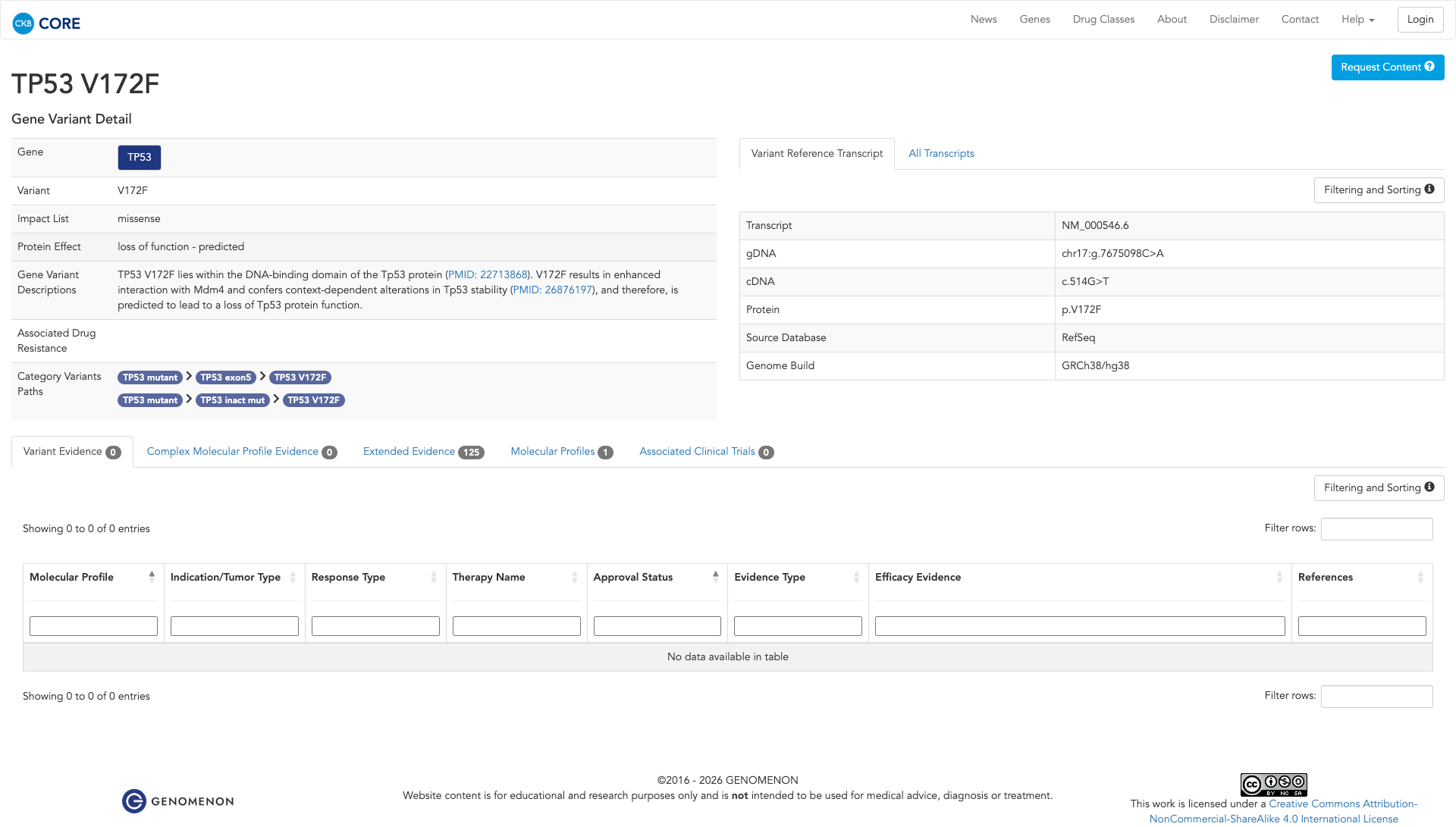
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | -144 bp |
| Donor Loss (DL) | 0.01 | 1 bp |
| Acceptor Gain (AG) | 0.0 | -127 bp |
| Donor Gain (DG) | 0.0 | 238 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, PVS1 applies to null variants (nonsense, frameshift, canonical ±1,2 splice sites, initiation codon) predicted to undergo NMD. The evidence for this variant shows it is a missense change (V172F). Therefore, this criterion is not applied.
PS1 (Not Applied)
According to standard ACMG guidelines, PS1 is "Same amino acid change as a previously established pathogenic variant regardless of nucleotide change." The evidence for this variant shows no other nucleotide change resulting in V172F has been classified as pathogenic. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, PS2 is "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history." No parental testing or de novo evidence is available. Therefore, this criterion is not applied.
PS3 (Strong)
According to standard ACMG guidelines, PS3 is "Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product." The evidence for this variant shows multiple functional assays demonstrating loss of transactivation activity and disrupted TP53 function. Therefore, this criterion is applied at Strong strength because well-established functional studies support damaging effect.
PS4 (Not Applied)
According to standard ACMG guidelines, PS4 is "The prevalence of the variant in affected individuals is significantly increased compared to controls." No case-control or proband observation data meeting the VCEP point thresholds are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to VCEP guidelines, PM1 (Moderate) applies to missense variants at TP53 codons 175, 245, 248, 249, 273, or 282. The evidence for this variant shows it affects codon 172, which is not among the specified hotspots. Therefore, this criterion is not applied.
PM2 (Supporting)
According to VCEP guidelines, PM2_Supporting is "Allele frequency <0.00003 in gnomAD or another large population database." The evidence for this variant shows it is absent from gnomAD. Therefore, this criterion is applied at Supporting strength because the variant is absent from controls.
PM3 (Not Applied)
According to standard ACMG guidelines, PM3 is "Detected in trans with a pathogenic variant for a recessive disorder." This is a dominant cancer predisposition gene and no trans data are relevant. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 is "Protein length changes due to in-frame deletions/insertions or stop-loss." The evidence for this variant shows a single amino acid substitution with no length change. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to VCEP guidelines, PM5 (Moderate) applies when another pathogenic missense variant at the same codon has been established. No other pathogenic variant at codon 172 is reported. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, PM6 is "Assumed de novo, but without confirmation of paternity and maternity." No de novo observations are reported. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to VCEP guidelines, PP1 requires cosegregation in affected family members. No segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, PP2 is "Missense variant in a gene with low rate of benign missense variation and where missense is a common mechanism." TP53 has many benign and pathogenic missense variants. Therefore, this criterion is not applied.
PP3 (Supporting)
According to standard ACMG guidelines, PP3 is "Multiple lines of computational evidence support a deleterious effect on the gene or gene product." The evidence for this variant shows a REVEL score of 0.92 and other in silico tools predict damaging. Therefore, this criterion is applied at Supporting strength because computational evidence supports deleterious effect.
PP4 (Not Applied)
According to standard ACMG guidelines, PP4 is "Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology." No detailed phenotype/family history specificity is provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, PP5 is "Reputable source reports variant as pathogenic but evidence not available." ClinVar reports are conflicting (Likely Pathogenic, VUS, Pathogenic). Due to conflicting assertions, this criterion is not applied.
BA1 (Not Applied)
According to standard ACMG guidelines, BA1 is "Allele frequency ≥0.001 in a large population database for a benign stand-alone rule." The variant is absent from gnomAD. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to standard ACMG guidelines, BS1 is "Allele frequency greater than expected for disorder but below BA1 threshold." The variant is absent from controls. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to VCEP guidelines, BS2 requires observation in ≥8 unaffected older females. No such data are available. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, BS3 is "Functional on Kato data AND no LOF on other assay." The variant shows loss of function, not preserved function. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to VCEP guidelines, BS4 requires lack of segregation in affected family members. No segregation data are available. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 is "Missense variant in a gene for which primarily truncating variants cause disease." TP53 disease mechanism is predominantly missense. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 is "Observed in trans with a pathogenic variant in dominant disorder or in cis with another pathogenic variant." No such phasing data are available. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 is "In-frame indel in repetitive region without known function." This is a missense variant, not an indel. Therefore, this criterion is not applied.
BP4 (Not Applied)
According to VCEP guidelines, BP4_Moderate is "BayesDel ≤ -0.008 and SpliceAI <0.2." Computational tools predict deleterious and SpliceAI not assessed for benign. Therefore, this criterion is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 is "Variant found in a case with an alternate molecular basis for disease." No such evidence is available. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 is "Reputable source reports variant as benign but evidence not available." No such benign assertions exist. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to VCEP guidelines, BP7 is for silent or intronic variants outside splice motifs. This is a missense change. Therefore, this criterion is not applied.