Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000249.3 | RefSeq Select | 2662 nt | 199–2469 |
| NM_000249.2 | Alternative | 2524 nt | 61–2331 |
| NM_000249.4 | MANE Select | 2494 nt | 31–2301 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenVariant summary: The MLH1 c.790+1G>A variant involves the alteration of a conserved intronic nucleotide. One in silico tool predicts a damaging outcome for this variant. 5/5 splice prediction tools predict that this variant ablishes the 5' splicing donor site, which was confirmed by an in vitro study (Auclair_2006). This variant has been found in numerous LS patients and is absent in 115338 control chromosomes. In addition, multiple clinical diagnostic laboratories/reputable databases classified this variant as pathogenic. Taken together, this variant is classified as pathogenic.
This variant is considered pathogenic. This variant occurs within a consensus splice junction and is predicted to result in abnormal mRNA splicing of either an out-of-frame exon or an in-frame exon necessary for protein stability and/or normal function. Functional studies indicate this variant impacts protein function [PMID: 8574961, 11781295].
The c.790+1G>A intronic pathogenic mutation results from a G to A substitution one nucleotide after coding exon 9 of the MLH1 gene. This mutation has been seen in multiple individuals with personal and family histories of colon cancer (Cunningham JM et al. Am. J. Hum. Genet. 2001 Oct;69:780-90; Rosty C et al. BMJ Open. 2016 Feb;6(2):e010293; Haraldsdottir S et al. Fam. Cancer 2016 Apr;15(2):253-60; Rossi BM et al. BMC Cancer 2017 Sep;17(1):623). In addition, a functional study utilizing RNA from an individual carrying this mutation found that it leads to skipping of coding exons 9 and 10 (Auclair J et al. Hum. Mutat. 2006 Feb;27:145-54; Ambry internal data). In addition to the clinical data presented in the literature, alterations that disrupt the canonical splice site are expected to cause aberrant splicing, resulting in an abnormal protein or a transcript that is subject to nonsense-mediated mRNA decay. As such, this alteration is classified as a disease-causing mutation.
The MLH1 c.790+1G>A variant (also known as IVS9+1G>A) disrupts a canonical splice site and has been shown to cause an in-frame skipping of exons 9-10 (PMID: 16395668 (2006)), at a level greater the alternatively spliced isoforms observed in normal cells (PMIDs: 9490293 (1998), 7728749 (1995)). The frequency of this variant in the general population, 0.0000066 (1/152090 chromosomes, http://gnomad.broadinstitute.org), is consistent with pathogenicity. The variant has been reported in multiple individuals with colorectal cancer or Lynch syndrome (PMIDs: 26895986 (2016), 25133505 (2014), 25107687 (2014), 24344984 (2013), 20937110 (2010), 20305446 (2010), 17054581 (2006), 16395668 (2006), 15955785 (2005), 15849733 (2005), 11524701 (2001)). A functional study also indicates the variant causes a significant loss of DNA mismatch repair activity (PMID: 11781295 (2002)). Based on the available information, this variant is classified as pathogenic.
"This variant has been reported in ClinVar as Pathogenic (17 clinical laboratories) and as Likely pathogenic (1 clinical laboratories) and as Pathogenic by International Society for Gastrointestinal Hereditary Tumours (InSiGHT) expert panel."
COSMIC Somatic Evidence
Open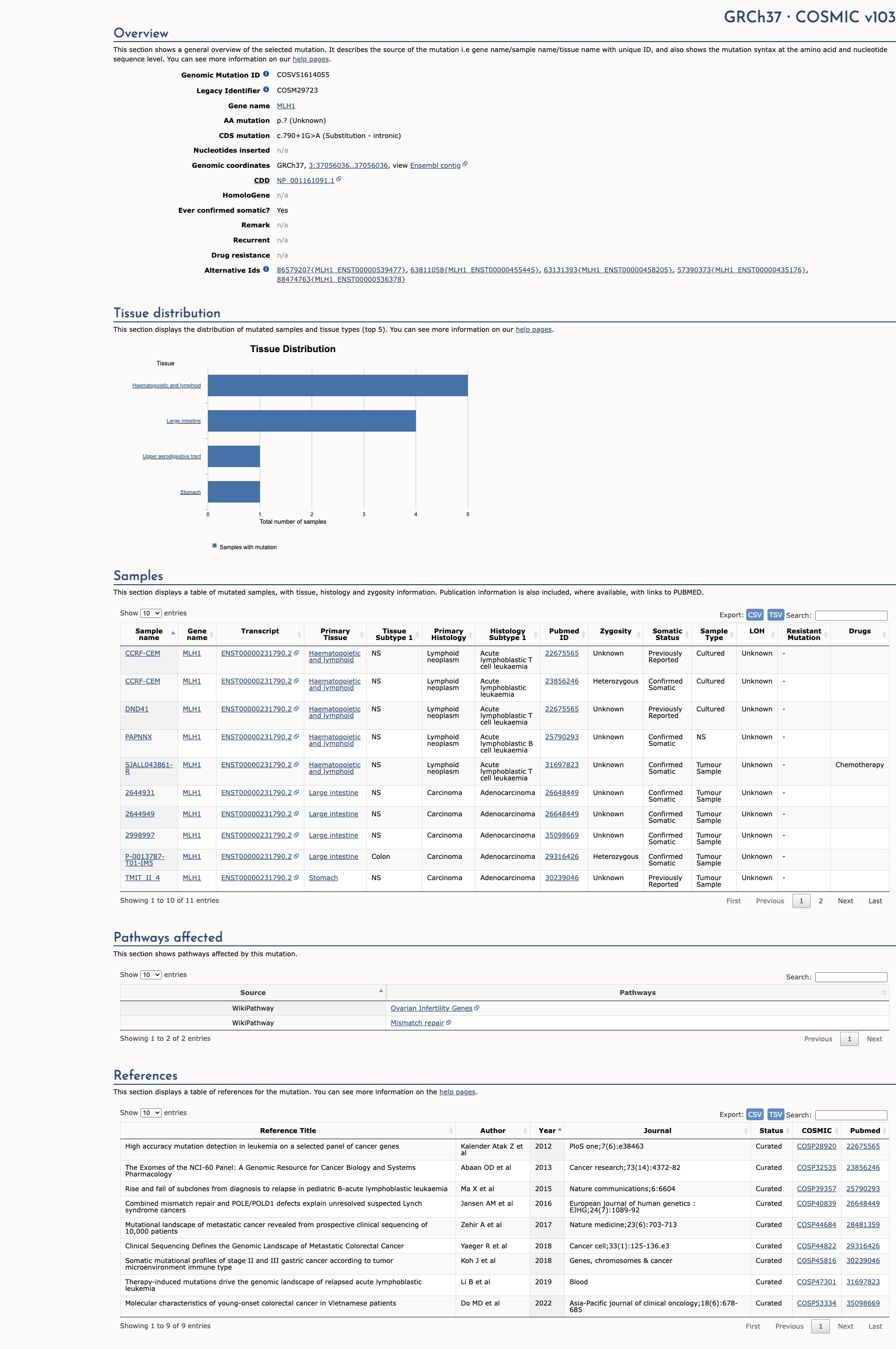
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.76 | -113 bp |
| Donor Loss (DL) | 0.87 | -1 bp |
| Acceptor Gain (AG) | 0.0 | 38 bp |
| Donor Gain (DG) | 0.0 | 89 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to VCEP guidelines, the rule for PVS1 Very Strong is: "Variants at IVS±1 or IVS±2 where exon skipping or use of a cryptic splice site disrupts reading frame and is predicted to undergo NMD." The evidence for this variant shows a canonical +1 splice site change (c.790+1G>A) in MLH1, predicted to abolish normal splicing and cause loss of function via NMD. Therefore, this criterion is applied at Very Strong strength because it meets the VCEP definition for a canonical splice disrupting variant predicted to lead to NMD.
PS1 (Not Applied)
According to VCEP guidelines, the rule for PS1 Strong is: "Variants affecting the same non-canonical splice nucleotide as a confirmed pathogenic splice variant with similar or worse splicing in silico prediction using SpliceAI." The evidence for this variant shows it is at the canonical +1 splice site, not a non-canonical nucleotide, and there is no prior non-canonical splice nucleotide variant at this exact position. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to VCEP guidelines, the rule for PS2 is de novo occurrence points: Very Strong ≥4, Strong 2–3, Moderate 1, Supporting 0.5. There is no information regarding de novo status. Therefore, this criterion is not applied.
PS3 (Not Applied)
According to VCEP guidelines, the rule for PS3 Strong is: "Calibrated functional assays with functional odds for Pathogenicity >18.7." No functional assays have been performed for this splice variant. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "Prevalence of variant in affected individuals statistically increased over controls." No case-control or segregation data are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot and/or critical and well‐established functional domain without benign variation." This variant affects splicing, not a known functional domain hotspot. Therefore, this criterion is not applied.
PM2 (Supporting)
According to VCEP guidelines, the rule for PM2 Supporting is: "Absent/extremely rare (<1 in 50,000 alleles) in gnomAD v4." The evidence for this variant shows MAF=0% in gnomAD v4. Therefore, this criterion is applied at Supporting strength because the variant is absent in population databases.
PM3 (Not Applied)
According to VCEP guidelines, the rule for PM3 applies to recessive disorders with trans observations. No trans occurrences with a pathogenic MLH1 variant have been reported. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame indels or stop-loss variants." This is a splice site variant leading to LOF covered by PVS1. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to VCEP guidelines, the rule for PM5 Moderate is: "Missense change at an amino acid residue where a different missense change was classified as Pathogenic." This variant is a splice site variant, not a missense change. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Unconfirmed de novo occurrence." No de novo evidence is available. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to VCEP guidelines, the rule for PP1 Strong is: "Co-segregation with disease with combined Bayes LR >18.7." No segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with low rate of benign missense variation." This is a splice variant, not missense. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to VCEP guidelines, the rule for PP3 Supporting is: "Predicted splice defect for non-canonical splicing nucleotides using SpliceAI with delta score ≥0.2." This is a canonical +1 splice variant for which PVS1 applies and VCEP specifies not to combine PP3 with PVS1. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to VCEP guidelines, the rule for PP4 Strong is: "Phenotype or family history specific for MLH1 pathogenic variants (e.g., MSI-H tumors)." No tumor phenotype data are provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as pathogenic without accessible evidence." Although ClinVar entries exist, VCEP does not recommend PP5. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 Stand Alone is: "gnomAD v4 Grpmax filtering allele frequency ≥0.001." The variant has MAF=0% in gnomAD. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to VCEP guidelines, the rule for BS1 Strong is: "gnomAD v4 Grpmax filtering allele frequency ≥0.0001 and <0.001." The variant has MAF=0% in gnomAD. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to VCEP guidelines, the rule for BS2 Strong is: "Co-occurrence in trans with a known pathogenic MLH1 variant in non-CMMRD context." No such co-occurrence data exist. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, the rule for BS3 Strong is: "Calibrated assays showing no functional impact." No functional assays demonstrate normal splicing or function. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to VCEP guidelines, the rule for BS4 Strong is: "Lack of segregation with disease (Bayes LR <0.05)." No segregation data are available. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where truncating variants are the predominant mechanism." This is a splice variant causing truncation, not missense. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for a dominant disorder." No such observation is reported. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame indels in repetitive regions without known function." This variant is a splice site change, not an in-frame indel. Therefore, this criterion is not applied.
BP4 (Not Applied)
According to VCEP guidelines, the rule for BP4 Supporting is: "SpliceAI predicts no splicing impact with delta score ≤0.1." SpliceAI predicts high impact (0.87). Therefore, this criterion is not applied.
BP5 (Not Applied)
According to VCEP guidelines, the rule for BP5 Strong is: "Tumor characteristics inconsistent with MLH1 loss." No such tumor data exist. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign without accessible evidence." No such benign reports exist. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to VCEP guidelines, the rule for BP7 Supporting is: "Synonymous or intronic variant at or beyond –21/+7." This is a canonical +1 variant. Therefore, this criterion is not applied.