Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000051.3 | RefSeq Select | 13147 nt | 386–9556 |
| NM_000051.4 | MANE Select | 12915 nt | 151–9321 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe p.M3011V variant (also known as c.9031A>G), located in coding exon 62 of the ATM gene, results from an A to G substitution at nucleotide position 9031. The methionine at codon 3011 is replaced by valine, an amino acid with highly similar properties. This variant has been identified in multiple individuals diagnosed with breast cancer (Teraoka SN et al. Cancer. 2001 Aug;92(3):479-87; Tavtigian SV et al. Am. J. Hum. Genet. 2009 Oct;85(4):427-46). Another study found this alteration in 1 of 270 breast and/or ovarian cancer families (Thorstenson YR et al. Cancer Res. 2003 Jun;63(12):3325-33). This alteration was also reported in 1/5560 prostate cancer cases and in 0/3353 controls of European ancestry (Karlsson Q et al. Eur Urol Oncol, 2021 08;4:570-579). This amino acid position is not well conserved in available vertebrate species. In addition, this alteration is predicted to be tolerated by in silico analysis. Based on the available evidence, the clinical significance of this variant remains unclear.
The ATM c.9031A>G (p.Met3011Val) variant has been reported in the published literature in individuals affected with breast cancer (PMID: 11505391 (2001), 12810666 (2003), 19781682 (2009)), glioblastoma (PMID: 26689913 (2015)), prostate cancer (PMID: 33436325 (2021)), and melanoma (PMID: 34262154 (2021)). This variant has also been identified in reportedly healthy individuals (PMID: 33471991 (2021), see also LOVD (http://databases.lovd.nl/shared/)). The frequency of this variant in the general population, 0.000077 (10/129144 chromosomes (Genome Aggregation Database, http://gnomad.broadinstitute.org)), is uninformative in the assessment of its pathogenicity. Analysis of this variant using bioinformatics tools for the prediction of the effect of amino acid changes on protein structure and function yielded conflicting predictions that this variant is deleterious or benign. Based on the available information, we are unable to determine the clinical significance of this variant.
Variant summary: ATM c.9031A>G (p.Met3011Val) results in a conservative amino acid change located in the Phosphatidylinositol 3-/4-kinase, catalytic domain (IPR000403) of the encoded protein sequence. Algorithms developed to predict the effect of missense changes on protein structure and function are either unavailable or do not agree on the potential impact of this missense change. The variant allele was found at a frequency of 4e-05 in 251442 control chromosomes. This frequency is not significantly higher than estimated for disease-causing variants in ATM, allowing no conclusion about variant significance. c.9031A>G has been reported in the literature in individuals affected with breast cancer (e.g. Tavtigian_2009, Teraoka_2001, Thorstenson_2003) and prostate cancer (e.g. Karlsson_2020) without strong evidence for causality. These reports do not provide unequivocal conclusions about association of the variant with Breast Cancer. At-least one co-occurrence with another pathogenic variant has been observed at our laboratory (APC c.487C>T, p.Gln163Ter), providing supporting evidence for a benign role. To our knowledge, no experimental evidence demonstrating an impact on protein function has been reported. The following publications have been ascertained in the context of this evaluation (PMID: 27200287, 30197789, 33436325, 26689913, 25148578, 19781682, 11505391, 12810666). ClinVar contains an entry for this variant (Variation ID: 181906). Based on the evidence outlined above, the variant was classified as VUS-possibly benign.
This sequence change replaces methionine, which is neutral and non-polar, with valine, which is neutral and non-polar, at codon 3011 of the ATM protein (p.Met3011Val). This variant is present in population databases (rs372795527, gnomAD 0.008%). This missense change has been observed in individual(s) with breast cancer, glioblastoma multiforme, prostate cancer, and pancreatic cancer (PMID: 11505391, 12810666, 19781682, 26689913, 33436325, 35047863). ClinVar contains an entry for this variant (Variation ID: 181906). Invitae Evidence Modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) indicates that this missense variant is not expected to disrupt ATM protein function with a negative predictive value of 95%. In summary, the available evidence is currently insufficient to determine the role of this variant in disease. Therefore, it has been classified as a Variant of Uncertain Significance.
"This variant has been reported in ClinVar as Uncertain significance (13 clinical laboratories) and as Likely benign (1 clinical laboratories)."
COSMIC Somatic Evidence
Open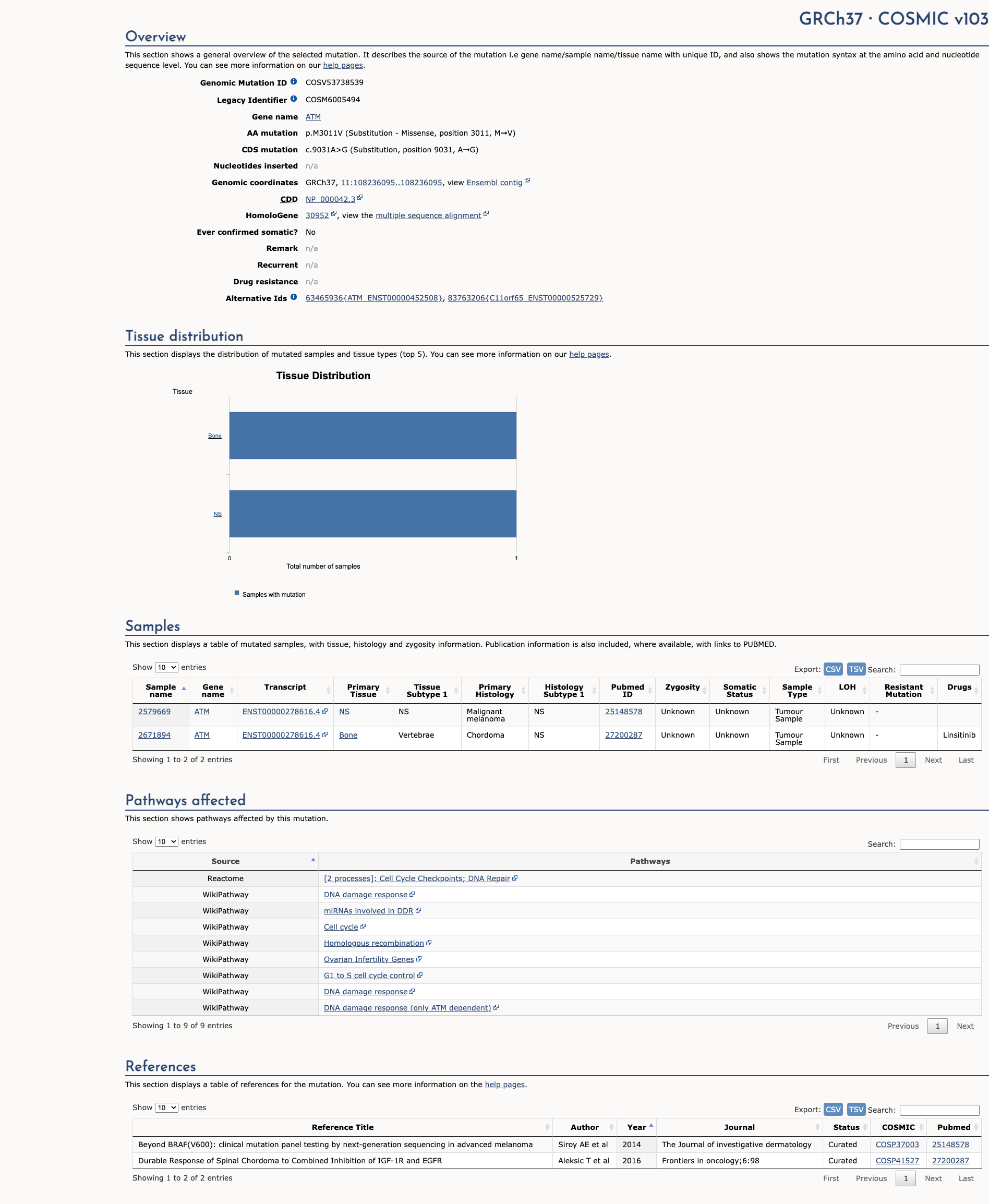
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.01 | -43 bp |
| Donor Loss (DL) | 0.01 | 134 bp |
| Acceptor Gain (AG) | 0.04 | 348 bp |
| Donor Gain (DG) | 0.0 | 50 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, the rule for PVS1 is: 'Very Strong Strength: Very Strong Use ATM PVS1 Decision Tree Modification Type: Gene-specific,Strength'. The evidence for this variant shows: NM_000051.4:c.9031A>G is a missense change (M3011V) and not a null variant. Therefore, this criterion is not applied because the variant does not meet any PVS1 null variant condition.
PS1 (Not Applied)
According to VCEP guidelines, the rule for PS1 is: 'Strong Strength: Strong Use for protein changes as long as splicing is ruled-out for both alterations. Use ATM PS1 Splicing table for splicing variants with similar predictions or observations of splice defect.'. The evidence for this variant shows: there is no previously established pathogenic variant with the same amino acid change at position M3011. Therefore, this criterion is not applied because the variant does not match any known pathogenic amino acid substitution.
PS2 (Not Applied)
According to standard ACMG guidelines, the rule for PS2 is: 'De novo (both maternity and paternity confirmed) in a patient with the disease and no family history'. The evidence for this variant shows: no data on de novo occurrence or parental testing. Therefore, this criterion is not applied due to lack of de novo confirmation.
PS3 (Not Applied)
According to VCEP guidelines, the rule for PS3 (Moderate Strength) is: 'Moderate Use when a variant fails to rescue both an ATM specific feature (e.g. phosphorylation of ATM-specific targets) AND radiosensitivity.'. The evidence for this variant shows: no functional studies have been performed. Therefore, this criterion is not applied because required functional assay evidence is missing.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: 'The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls (e.g., case-control data).'. The evidence for this variant shows: no case-control or proband counting data. Therefore, this criterion is not applied due to absence of statistical evidence.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: 'Located in a mutational hot spot and/or critical and well-established functional domain without benign variation.'. The evidence for this variant shows: M3011V is not reported in a known functional domain hotspot in ATM. Therefore, this criterion is not applied.
PM2 (Not Applied)
According to VCEP guidelines, the rule for PM2 (Supporting Strength) is: 'Frequency ≤.001% if n=1 in a single sub population; n>1 in one or multiple subpopulations would not be considered rare and PM2_supporting would not apply.'. The evidence for this variant shows: allele count is 11/282,840 (MAF=0.00389%), n>1 and >0.001%. Therefore, this criterion is not applied.
PM3 (Not Applied)
According to VCEP guidelines, the rule for PM3 involves using the ATM PM3/BP2 table for recessive in trans observations. The evidence for this variant shows: no data on trans configuration with a pathogenic variant. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: 'Protein length changes due to in-frame deletions/insertions in a non-repeat region or stop-loss variants.'. The evidence for this variant shows: M3011V is a missense substitution and does not alter protein length. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to VCEP guidelines, the rule for PM5 (Supporting Strength) is: 'Supporting Use for genomic frameshift and truncating variants with PTC upstream of p.R3047...'. The evidence for this variant shows: M3011V is a missense change, not a truncating variant. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: 'Assumed de novo (without confirmation of paternity and maternity).'. The evidence for this variant shows: no parental relationship information. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, the rule for PP1 is: 'Co-segregation with disease in multiple affected family members.'. The evidence for this variant shows: no familial segregation data. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: 'Missense variant in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease.'. The evidence for this variant shows: ATM has a higher rate of tolerated missense and disease is typically caused by loss-of-function rather than missense. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to VCEP guidelines, the rule for PP3 (Supporting Strength) is: 'Supporting Protein: REVEL >.7333; RNA: At least one well-established in silico predictor shows impact on splicing.'. The evidence for this variant shows: REVEL=0.18 (<0.7333) and SpliceAI=0.04 (no splicing impact). Therefore, this criterion is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 is: 'Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology.'. The evidence for this variant shows: no phenotype or clinical presentation provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: 'Reputable source reports variant as pathogenic.'. The evidence for this variant shows: ClinVar submissions are predominantly VUS with one Likely Benign; no reputable source labeled it pathogenic. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 (Stand Alone Strength) is: 'Filtering Allele Frequency >.5%'. The evidence for this variant shows: MAF=0.00389% which is <<0.5%. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to VCEP guidelines, the rule for BS1 (Strong Strength) is: 'Filtering Allele Frequency >.05%'. The evidence for this variant shows: MAF=0.00389% which is <0.05%. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 is: 'Observed in a healthy adult individual for a dominant (with full penetrance expected at an early age), X-linked or mitochondrial disorder with full penetrance expected at an early age.'. The evidence for this variant shows: no healthy adult observation data. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, the rule for BS3 (Moderate Strength) is: 'Use when a variant rescues both an ATM specific feature (e.g. phosphorylation of ATM-specific targets) AND radiosensitivity.'. The evidence for this variant shows: no functional rescue data. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: 'Lack of segregation in affected members of a family.'. The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: 'Missense variant in a gene for which primarily truncating variants are known to cause disease.'. The evidence for this variant shows: ATM missense variants can be pathogenic. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to VCEP guidelines, the rule for BP2 involves the ATM PM3/BP2 table for cis observations. The evidence for this variant shows: no data on cis with pathogenic variant. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: 'In-frame deletions/insertions in a repetitive region without a known function.'. The evidence for this variant shows: M3011V is a missense substitution, not an indel. Therefore, this criterion is not applied.
BP4 (Supporting)
According to VCEP guidelines, the rule for BP4 (Supporting Strength) is: 'Supporting Protein Analysis: Metapredictor REVEL score ≤.249; RNA: At least one well-established in silico predictor (e.g. SpliceAI) shows impact on splicing; NOTE: Splice analysis needs to be considered for all variant types...'. The evidence for this variant shows: REVEL=0.18 (≤.249) and SpliceAI max=0.04 (no splicing impact). Therefore, this criterion is applied at Supporting strength because both protein and RNA computational predictors support a benign effect.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: 'Variant found in a case with an alternate molecular basis for disease.'. The evidence for this variant shows: no such case data. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: 'Reputable source reports variant as benign.'. The evidence for this variant shows: ClinVar entries include one Likely Benign but not sufficient to be considered a well-established reputable source. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 is: 'Synonymous variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor creation of a new splice site.'. The evidence for this variant shows: it is a missense, not synonymous. Therefore, this criterion is not applied.