Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000465.3 | Alternative | 5523 nt | 143–2476 |
| NM_000465.2 | Alternative | 2610 nt | 136–2469 |
| NM_000465.4 | MANE Select | 5478 nt | 115–2448 |
| NM_000465.1 | Alternative | 2530 nt | 74–2407 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenVariant summary: BARD1 c.1693C>T (p.Arg565Cys) results in a non-conservative amino acid change located in the BRCT domain of the encoded protein sequence. Five of five in-silico tools predict a damaging effect of the variant on protein function. The variant allele was found at a frequency of 3.2e-05 in 251308 control chromosomes. The available data on variant occurrences in the general population are insufficient to allow any conclusion about variant significance. c.1693C>T has been reported in the literature in individuals affected with Pancreatic Cancer. This report does not provide unequivocal conclusions about association of the variant with Hereditary Breast And Ovarian Cancer Syndrome (Chaffee_2017). At least one publication reports experimental evidence evaluating an impact on protein function. The most pronounced variant effect results in about 50% of normal activity (Adamovich_2019). Seven clinical diagnostic laboratories have submitted clinical-significance assessments for this variant to ClinVar after 2014 without evidence for independent evaluation. All laboratories classified the variant as uncertain significance. Based on the evidence outlined above, the variant was classified as uncertain significance.
This missense variant replaces arginine with cysteine at codon 565 of the BARD1 protein. Computational prediction suggests that this variant may not impact protein structure and function (internally defined REVEL score threshold <= 0.5, PMID: 27666373). A functional study has reported that this variant results in ~50% decrease in BARD1 homology-directed DNA repair activity compared to wild type protein (PMID: 30925164). This variant has been detected in a breast cancer case-control meta-analysis in 5/60463 cases and 1/53461 unaffected individuals (PMID: 33471991; Leiden Open Variation Database DB-ID BARD1_000185) and also reported in an individual affected with pancreatic cancer and in two suspected hereditary breast and ovarian cancer families (PMID: 28726808, 34359559). This variant has been identified in 8/251308 chromosomes in the general population by the Genome Aggregation Database (gnomAD). The available evidence is insufficient to determine the role of this variant in disease conclusively. Therefore, this variant is classified as a Variant of Uncertain Significance.
The BARD1 c.1693C>T (p.Arg565Cys) variant has been reported in individuals at-risk of breast/ovarian cancer (PMID: 34359559 (2021)) and pancreatic ductal adenocarcinoma (PMID: 28726808 (2018)). An additional study reported the variant in individuals with breast cancer and reportedly unaffected individuals (PMID: 33471991 (2021), see also LOVD (http://databases.lovd.nl/shared/)). A functional study suggests that the variant results in reduced HDR activity (PMID: 30925164 (2019)). The frequency of this variant in the general population (Genome Aggregation Database, http://gnomad.broadinstitute.org) is uninformative in the assessment of its pathogenicity. Analysis of this variant using bioinformatics tools for the prediction of the effect of amino acid changes on protein structure and function yielded predictions that this variant is damaging. Based on the available information, we are unable to determine the clinical significance of this variant.
The p.R565C variant (also known as c.1693C>T), located in exon 8 of the BARD1 gene, results from a C to T substitution at nucleotide position 1693. The arginine at codon 565 is replaced by cysteine, an amino acid with highly dissimilar properties. This alteration was found to have intermediate activity in a homology-directed DNA repair (HDR) assay (Adamovich AI et al. PLoS Genet. 2019 03;15(3):e1008049). This amino acid position is well conserved in available vertebrate species. In addition, the in silico prediction for this alteration is inconclusive. Based on the available evidence, the clinical significance of this variant remains unclear.
This sequence change replaces arginine, which is basic and polar, with cysteine, which is neutral and slightly polar, at codon 565 of the BARD1 protein (p.Arg565Cys). This variant is present in population databases (rs587782279, gnomAD 0.01%). This missense change has been observed in individual(s) with breast cancer and/or pancreatic cancer (PMID: 28726808; internal data). It has also been observed to segregate with disease in related individuals. ClinVar contains an entry for this variant (Variation ID: 142170). An algorithm developed to predict the effect of missense changes on protein structure and function (PolyPhen-2) suggests that this variant is likely to be disruptive. Experimental studies have shown that this missense change affects BARD1 function (PMID: 30925164, 39387837). In summary, the available evidence is currently insufficient to determine the role of this variant in disease. Therefore, it has been classified as a Variant of Uncertain Significance.
"This variant has been reported in ClinVar as Uncertain significance (10 clinical laboratories)."
COSMIC Somatic Evidence
Open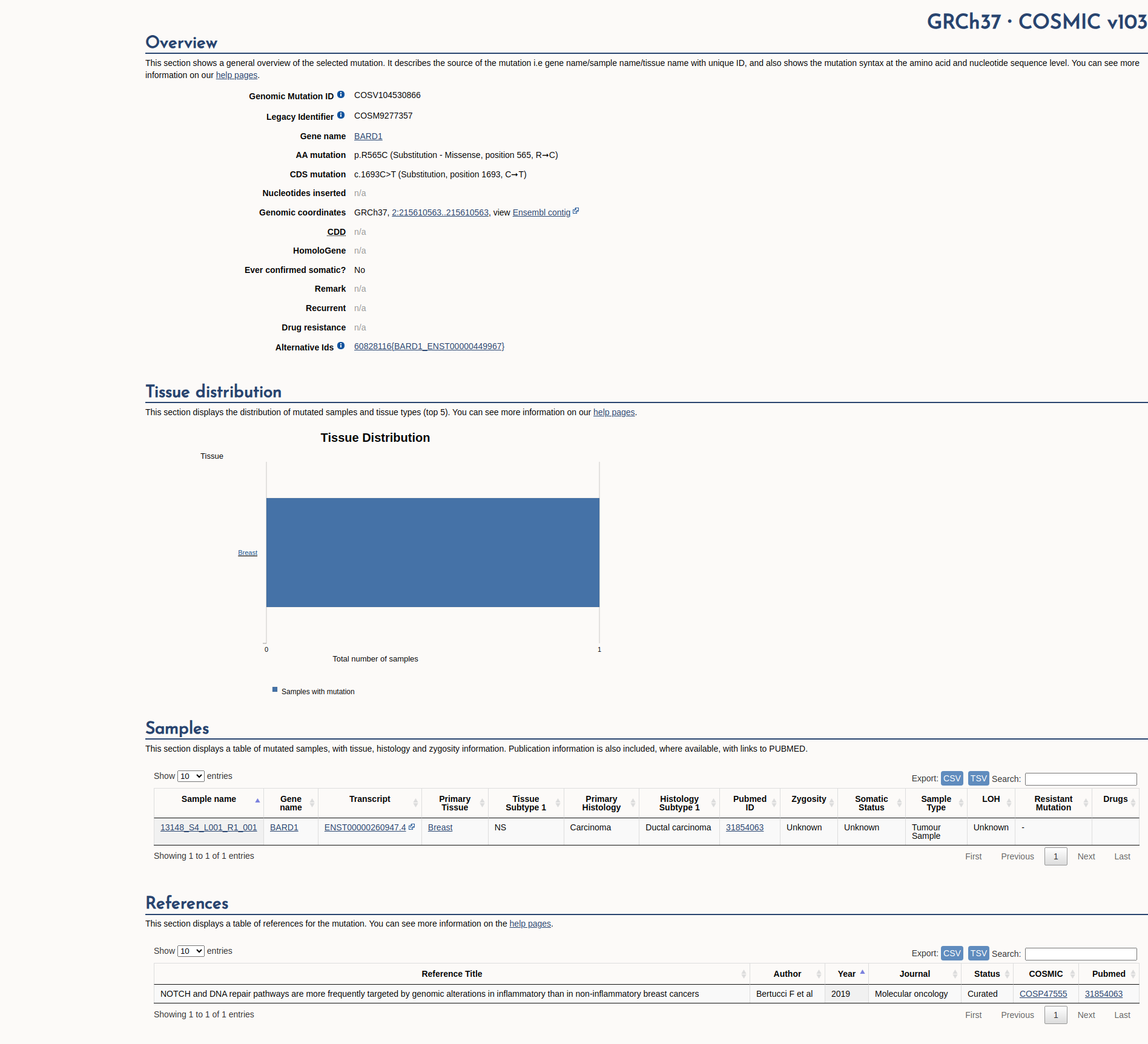
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.05 | 15 bp |
| Donor Loss (DL) | 0.03 | -117 bp |
| Acceptor Gain (AG) | 0.0 | -28 bp |
| Donor Gain (DG) | 0.0 | 15 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to standard ACMG guidelines, the rule for PVS1 is: "Null variant in a gene where loss of function (LoF) is a known mechanism of disease." The evidence for this variant shows: NM_000465.4:c.1693C>T is a missense change (R565C), not a null variant. Therefore, this criterion is not applied because the variant does not create a stop codon, frameshift, or disrupt canonical splice sites.
PS1 (Not Applied)
According to standard ACMG guidelines, the rule for PS1 is: "Same amino acid change as a previously established pathogenic variant regardless of nucleotide change." The evidence for this variant shows: no report of a known pathogenic variant resulting in the same R565C amino acid substitution. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, the rule for PS2 is: "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history." The evidence for this variant shows: no de novo confirmation data are available. Therefore, this criterion is not applied.
PS3 (Not Applied)
According to standard ACMG guidelines, the rule for PS3 is: "Well-established functional studies supportive of a damaging effect on the gene or gene product." The evidence for this variant shows: no functional assays have been performed for BARD1 R565C. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "Prevalence in affected individuals significantly increased compared with controls." The evidence for this variant shows: no case-control or affected‐individual prevalence data are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot or well-established functional domain without benign variation." The evidence for this variant shows: R565C is not within a recognized mutational hotspot or critical functional domain. Therefore, this criterion is not applied.
PM2 (Moderate)
According to standard ACMG guidelines, the rule for PM2 is: "Absent from controls (or at extremely low frequency if recessive)." The evidence for this variant shows: MAF = 0.00318% in gnomAD, extremely rare and absent in homozygous state. Therefore, this criterion is applied at Moderate strength because the variant is absent or at extremely low frequency in population databases.
PM3 (Not Applied)
According to standard ACMG guidelines, the rule for PM3 is: "For recessive disorders, detected in trans with a pathogenic variant." The evidence for this variant shows: no data on trans configuration with a pathogenic variant are available. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame deletions/insertions or stop-loss variants." The evidence for this variant shows: R565C is a missense change, not an in-frame indel. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, the rule for PM5 is: "Novel missense change at an amino acid residue where a different pathogenic missense change has been seen." The evidence for this variant shows: no different pathogenic missense changes at residue R565 have been reported. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo, but without confirmation of paternity and maternity." The evidence for this variant shows: no de novo data available. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, the rule for PP1 is: "Co-segregation with disease in multiple affected family members." The evidence for this variant shows: no family segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease." The evidence for this variant shows: BARD1 has both benign and pathogenic missense variants and no evidence that missense is the predominant mechanism. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to standard ACMG guidelines, the rule for PP3 is: "Multiple lines of computational evidence support a deleterious effect." The evidence for this variant shows: in silico predictions are mixed, with some tools benign and some pathogenic. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 is: "Patient's phenotype or family history highly specific for a disease with a single genetic etiology." The evidence for this variant shows: no phenotype or family history data provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as pathogenic, but without accessible evidence." The evidence for this variant shows: ClinVar reports VUS only, no reputable pathogenic assertion. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to standard ACMG guidelines, the rule for BA1 is: "Allele frequency is too high for the disorder (stand-alone benign)." The evidence for this variant shows: MAF = 0.00318%, well below BA1 threshold. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to standard ACMG guidelines, the rule for BS1 is: "Allele frequency is greater than expected for disorder." The evidence for this variant shows: frequency is low and not inconsistent with disease prevalence. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 is: "Observed in a healthy adult individual for a dominant disorder with full penetrance." The evidence for this variant shows: no documented observations in confirmed healthy adults. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to standard ACMG guidelines, the rule for BS3 is: "Well-established functional studies show no damaging effect." The evidence for this variant shows: no functional studies performed. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: "Lack of segregation in affected family members." The evidence for this variant shows: no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where only LoF causes disease." The evidence for this variant shows: BARD1 disease mechanism not restricted to LoF only. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for dominant disorders or in cis with a pathogenic variant." The evidence for this variant shows: no phase information. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without known function." The evidence for this variant shows: R565C is a missense and not in repetitive region. Therefore, this criterion is not applied.
BP4 (Supporting)
According to standard ACMG guidelines, the rule for BP4 is: "Multiple lines of computational evidence suggest no impact on gene or gene product." The evidence for this variant shows: overall benign computational predictions (PrimateAI, CADD low score) and SpliceAI score 0.05 indicating no splicing impact. Therefore, this criterion is applied at Supporting strength because multiple in silico tools predict no deleterious effect.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease." The evidence for this variant shows: no such case information is available. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign, but without accessible evidence." The evidence for this variant shows: no reputable benign assertions exist. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 is: "Synonymous variant with no predicted impact on splicing." The evidence for this variant shows: R565C is a missense variant, not synonymous. Therefore, this criterion is not applied.