Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000249.4 | MANE Select | 2494 nt | 31–2301 |
| NM_000249.3 | RefSeq Select | 2662 nt | 199–2469 |
| NM_000249.2 | Alternative | 2524 nt | 61–2331 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenVariant summary: MLH1 c.649C>T (p.Arg217Cys) results in a non-conservative amino acid change located in the N-terminal domain (IPR002099) of the encoded protein sequence. Five of five in-silico tools predict a damaging effect of the variant on protein function. 4/4 computational tools predict no significant impact on normal splicing. The variant allele was found at a frequency of 0.00041 in 252977 control chromosomes, predominantly at a frequency of 0.0047 within the East Asian subpopulation in the gnomAD database. The observed variant frequency within East Asian control individuals in the gnomAD database is approximately 7 fold of the estimated maximal expected allele frequency for a pathogenic variant in MLH1 causing Hereditary Nonpolyposis Colorectal Cancer phenotype (0.00071), strongly suggesting that the variant is a benign polymorphism found primarily in populations of East Asian origin. c.649C>T has also been reported in the literature in several individuals affected with Lynch Syndrome, who were predominantly of Asian descent and the majority of these publications only performed evaluation of MLH1 and could not rule out variants in other Lynch syndrome associated genes. These reports therefore do not provide unequivocal conclusions about association of the variant with Lynch Syndrome. In addition, in one publication (Sapari 2014), the variant was found in co-occurrence with another pathogenic MLH1 variant, c.790+1G>A (although it was only identified through NGS and not confirmed by Sanger sequencing). One co-occurrence with another pathogenic variant has been reported internally (BRCA2 c.1648G>T , p.Glu550X), providing supporting evidence for a benign role. Several publications reported experimental evidence evaluating an impact on protein function (Ellison 2001, Trojan 2002, Kondo 2003, Takahashi 2007, Zhao 2008, Peng 2016), showing no or only a mild effect on function. A recent study suggested based on in vitro evidence that the variant might affect splicing by affecting splicesome assembly (Rhine 2018), however earlier reports that identified the variant in patients through RNA-based direct sequencing, did not indicate any specific splicing effect (Furukawa 2002, Wang 2005); therefore, the significance of this finding is uncertain. The following publications have been ascertained in the context of this evaluation (PMID: 12810663, 18373977, 11555625, 8797773, 18069769, 17510385, 9419403, 15342696, 8581513, 11781295, 15613555, 11920458, 24933000, 25882375, 21901500, 27553368, 22136435, 16425354, 27173243, 27093186, 25338684, 28767289, 29050249, 29505604, 30850667, 31666926). Nine submitters have cited clinical-significance assessments for this variant to ClinVar after 2014 and classified as benign/likely benign (n=7) and VUS (n=2). Based on the evidence outlined above, the variant was classified as benign.
This alteration is classified as likely benign based on a combination of the following: seen in unaffected individuals, population frequency, intact protein function, lack of segregation with disease, co-occurrence, RNA analysis, in silico models, amino acid conservation, lack of disease association in case-control studies, and/or the mechanism of disease or impacted region is inconsistent with a known cause of pathogenicity.
This variant is considered benign. This variant is strongly associated with less severe personal and family histories of cancer, typical for individuals without pathogenic variants in this gene [PMID: 27363726]. This variant has been observed at a population frequency that is significantly greater than expected given the associated disease prevalence and penetrance.
Variant classification was performed using the VarSome platform (https://varsome.com/). The variant meet BP6, BP4, PM2, and PP2. A number of high confidence submitters have classified as benign and likely benign. There is also no clinical evidence supporting a diagnosis of hereditary colorectal cancer. Assertion score is -4 according to PMID:32720330.
"This variant has been reported in ClinVar as Likely benign (7 clinical laboratories) and as Benign (5 clinical laboratories) and as Uncertain significance (2 clinical laboratories)."
COSMIC Somatic Evidence
Open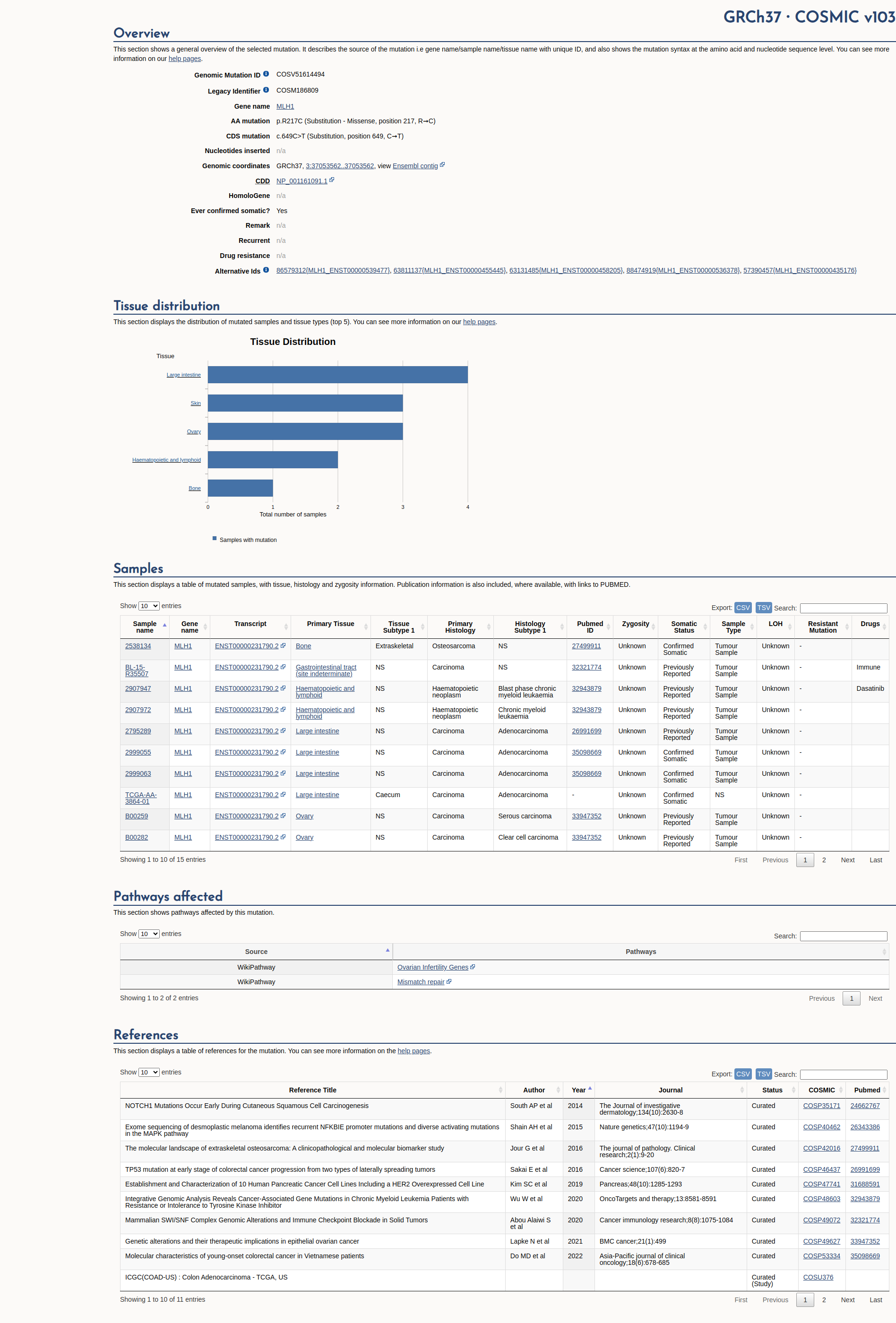
Functional Impact & Domains
Functional Domain
The MLH1 R217C variant has been functionally characterized with conflicting evidence. It demonstrates mismatch repair activity similar to wild-type MLH1 in cell-based assays, indicating a likely neutral effect. However, it also shows impaired interaction with PMS2, suggesting potential functional impairment. Overall, the effect on MLH1 protein function remains uncertain.
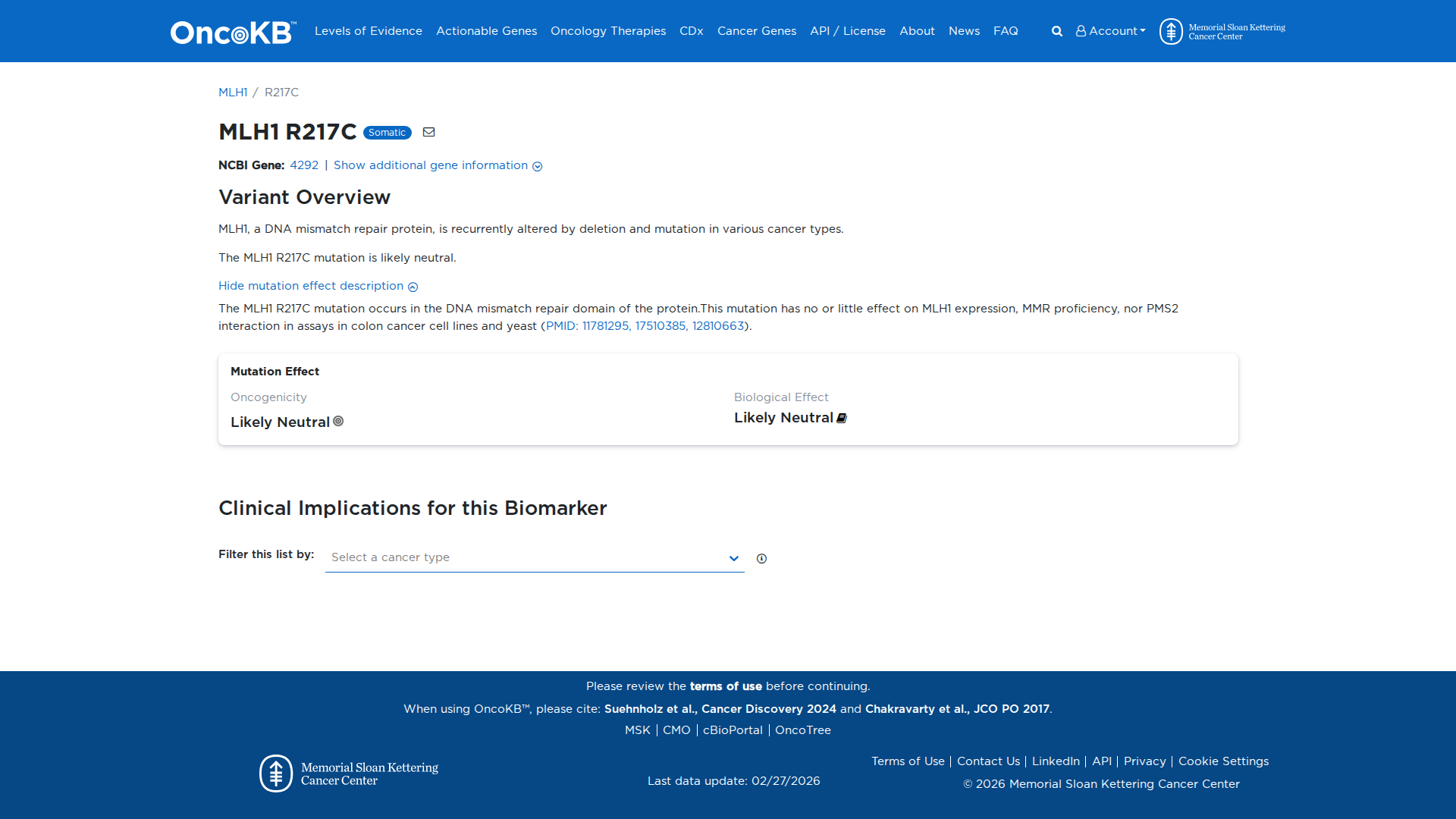
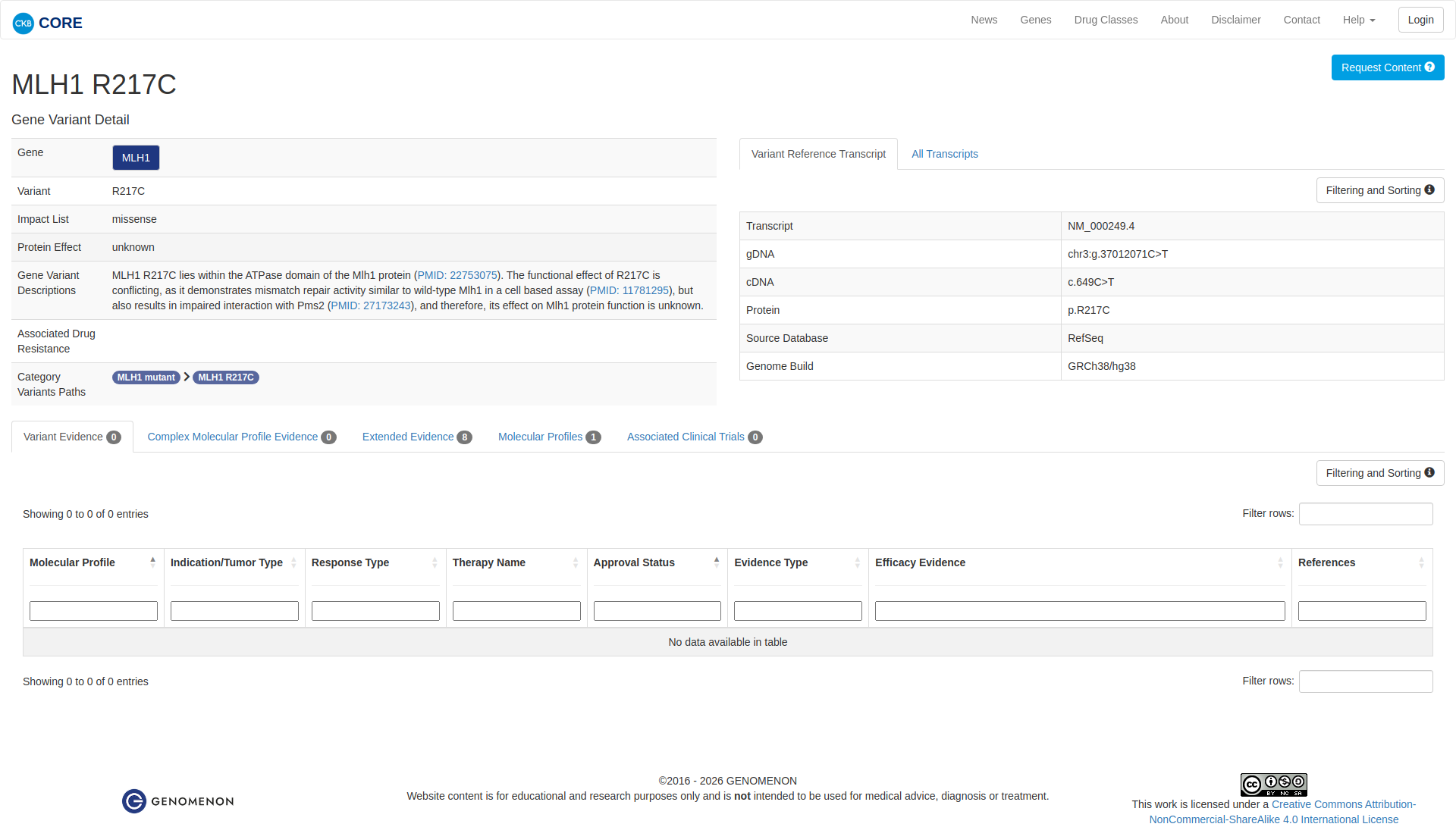
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.01 | -60 bp |
| Donor Loss (DL) | 0.01 | 28 bp |
| Acceptor Gain (AG) | 0.02 | -56 bp |
| Donor Gain (DG) | 0.0 | 24 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, the rule for PVS1 is: "Very Strong Strength: Very Strong Nonsense/frameshift variant introducing Premature Termination Codon (PTC) at or before codon 753 in MLH1." The evidence for this variant shows a missense substitution (R217C), not a null variant. Therefore, this criterion is not applied.
PS1 (Not Applied)
According to VCEP guidelines, the rule for PS1 is: "Strong: A predicted missense substitution that encodes the same amino acid change with a different underlying nucleotide change previously established by this VCEP as Pathogenic." The evidence for this variant shows no prior pathogenic R217C event at the amino acid level. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to VCEP guidelines, the rule for PS2 is: "Very Strong: ≥4 de novo points; Strong: 2–3 de novo points; Moderate: 1 de novo point; Supporting: 0.5 de novo points." The evidence for this variant shows no confirmed de novo occurrence. Therefore, this criterion is not applied.
PS3 (Not Applied)
According to VCEP guidelines, the rule for PS3 is: "Strong: Calibrated functional assays with functional odds for Pathogenicity >18.7; Moderate: odds >4.3 and ≤18.7 or MMR function defect; Supporting: odds >2.08 and ≤4.3." The evidence for this variant shows conflicting functional data (normal mismatch repair but impaired PMS2 interaction) without calibrated odds. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "The prevalence of the variant in affected individuals is significantly increased compared with controls." The evidence for this variant shows no case–control or cohort data. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Location in a mutational hot spot and/or critical and well‐established functional domain without benign variation." The evidence for this variant does not indicate a known mutational hotspot or critical domain at R217. Therefore, this criterion is not applied.
PM2 (Not Applied)
According to VCEP guidelines, the rule for PM2 (Supporting) is: "Absent/extremely rare (<1 in 50,000 alleles) in gnomAD v4." The evidence for this variant shows a gnomAD MAF of 0.034% (1 in ~2,941 alleles), which exceeds the threshold. Therefore, this criterion is not applied.
PM3 (Not Applied)
According to VCEP guidelines, the rule for PM3 is: "Points-based evaluation for variants in trans with a known pathogenic variant in recessive disease." The evidence for this variant shows no trans observations in a recessive context for MLH1. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in‐frame deletions/insertions or stop-loss variants." The evidence for this variant shows a missense change without length alteration. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to VCEP guidelines, the rule for PM5 is: "Moderate: Missense change at a residue where a different missense change was classified as Pathogenic by this VCEP; Supporting if the other is Likely Pathogenic." The evidence for this variant shows no other missense at R217 classified as Pathogenic. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo without confirmation of paternity/maternity." The evidence for this variant shows no de novo data. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to VCEP guidelines, the rule for PP1 is: "Supporting/Moderate/Strong based on segregation Bayes LR thresholds." The evidence for this variant shows no segregation data in families. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with low rate of benign missense variation and where missense is a common mechanism of disease." MLH1 has known pathogenic and benign missense variants; without constraint metrics, this cannot be applied. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to VCEP guidelines, the rule for PP3 is: "Supporting: HCI prior >0.68 & ≤0.81 or predicted splice defect (SpliceAI ≥0.2); Moderate: HCI prior >0.81." The evidence for this variant shows mixed in silico predictions and SpliceAI score 0.02 (<0.2). Therefore, this criterion is not applied.
PP4 (Not Applied)
According to VCEP guidelines, the rule for PP4 is: "Supporting/Moderate/Strong based on number of independent MSI-H tumors with loss of MMR expression." The evidence for this variant shows no tumor phenotype or MSI data. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as Pathogenic." The evidence for this variant shows only benign or VUS assertions in ClinVar, no pathogenic assertions. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 is: "Stand Alone: gnomAD v4 Grpmax filtering allele frequency ≥0.001 (0.1%)." The evidence for this variant shows MAF 0.00034 (<0.001). Therefore, this criterion is not applied.
BS1 (Strong)
According to VCEP guidelines, the rule for BS1 is: "Strong: gnomAD v4 Grpmax filtering allele frequency ≥0.0001 and <0.001." The evidence for this variant shows MAF 0.00034, which falls within this range. Therefore, this criterion is applied at Strong strength.
BS2 (Not Applied)
According to VCEP guidelines, the rule for BS2 is: "Strong: Co-occurrence in trans with a known pathogenic variant in a patient without clinical manifestations of CMMRD." The evidence for this variant shows no such co-occurrence data. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, the rule for BS3 is: "Strong: Calibrated functional assays with odds ≤0.05 or synonymous/intronic with no mRNA aberration; Supporting: odds >0.05 & ≤0.48 or proficient function." The evidence for this variant shows conflicting functional results without calibrated odds. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: "Strong: Lack of co-segregation with disease (Bayes LR <0.05); Supporting: Bayes LR >0.05 & ≤0.48." The evidence for this variant shows no segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene for which primarily truncating variants cause disease." MLH1 has known pathogenic missense variants, so this rule does not apply. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for a dominant disorder or in cis with a pathogenic variant in any disorder." There is no evidence of such occurrence. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without a known function." The evidence for this variant shows a missense change, not an in-frame indel. Therefore, this criterion is not applied.
BP4 (Supporting)
According to VCEP guidelines, the rule for BP4 is: "Supporting: Missense variant with HCI-prior <0.11 or SpliceAI delta score ≤0.1." The evidence for this variant shows a SpliceAI maximum delta score of 0.02 (≤0.1) and mixed missense predictions. Therefore, this criterion is applied at Supporting strength.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease." There is no evidence of an alternate cause. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign without available evidence." While ClinVar contains benign assertions, they are not sufficiently documented under this criterion. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to VCEP guidelines, the rule for BP7 is: "Supporting: Synonymous or intronic variant at or beyond –21/+7 with no splicing impact." The evidence for this variant shows a missense change. Therefore, this criterion is not applied.