Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000251.3 | MANE Select | 3115 nt | 37–2841 |
| NM_000251.2 | RefSeq Select | 3226 nt | 126–2930 |
| NM_000251.1 | Alternative | 3145 nt | 69–2873 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.942+3A>T variant in MSH2 has been reported in >150 individuals with Lynch syndrome (LabCorp database, Woods 2010). This variant has been identified in 1/12862 of European chromosomes by gnomAD (https://gnomad.broadinstitute.org/). In vitro functional studies show that the c.942+3A>T variant leads to an in-frame deletion of exon 5 (Auclair 2006). In addition, it has been classified as Pathogenic on December 18, 2013 by the ClinGen-approved InSiGHT expert panel (ClinVar SCV000107794.2). In summary, this variant meets criteria to be classified as pathogenic for Lynch syndrome in an autosomal dominant manner based upon the predicted impact to the protein, low frequency in controls and presence in affected individuals. ACMG/AMP Criteria applied: PS4, PM2, PS3_Supporting, PVS1_Strong.
This variant causes an A to T nucleotide substitution at the +3 position of intron 5 of the MSH2 gene. RNA studies have shown this variant caused skipping of exon 5 (r.793_942del) and in-frame deletion of 50 amino acids in the DNA binding domain (PMID: 8062247, 16395668, 19267393). This variant has been reported as a recurrent de novo mutation in individuals affected with Lynch syndrome-associated cancer in different ethnicities (PMID: 10978353). This variant has been reported in many individuals and families with Lynch syndrome or suspected Lynch syndrome worldwide (PMID: 8062247, 10413423, 10446963, 11920650, 12112654, 12362047, 15222003, 16203774, 16395668, 17312306, 18625694, 19130300, 19419416, 20682701, 21681552, 22883484), and shown to segregate with Lynch syndrome cancers in family studies (PMID: 19267393). This variant is considered to be a founder mutation in the Newfoundland population (PMID: 20682701). This variant has been identified in 1/30582 chromosomes in the general population by the Genome Aggregation Database (gnomAD). Based on the available evidence, this variant is classified as Pathogenic.
The c.942+3A>T intronic pathogenic mutation results from an A to T substitution 3 nucleotides after coding exon 5 in the MSH2 gene. In one study, this mutation was found in eight unrelated Lynch syndrome families. Six of the eight families (75%) had one family member diagnosed with either a keratocanthoma or sebaceous adenoma associated with Muir-Torre syndrome, along with some combination of colon, uterine, and/or ureter transitional cell cancers in the family (South CD et al. J. Natl. Cancer Inst. 2008 Feb;100:277-81). This mutation was also reported in a German patient diagnosed with malignant fibrous histiocytoma (MFH) (Brieger A et al. Fam. Cancer. 2011 Sep;10:591-5). Authors of one study claim that this is the most common recurrent de novo germline mutation in a human mismatch repair gene, accounting for approximately 11% of all known pathogenic MSH2 gene mutations (Desai DC et al. J. Med. Genet. 2000 Sep;37:646-52). Multiple studies have demonstrated that this mutation results in an mRNA transcript lacking coding exon 5 (Casey G et al. JAMA. 2005 Feb;293:799-809; Auclair J et al. Hum. Mutat. 2006 Feb;27:145-54; Arnold S et al. Hum. Mutat. 2009 May;30:757-70; Chong G et al. Hum. Mutat. 2009 Aug;30:E797-812). Of note, this mutation is also designated as IVS5+3A>T in published literature. Based on the supporting evidence, this alteration is interpreted as a disease-causing mutation.
This variant causes an A to T nucleotide substitution at the +3 position of intron 5 of the MSH2 gene. RNA studies have shown this variant caused skipping of exon 5 (r.793_942del) and in-frame deletion of 50 amino acids in the DNA binding domain (PMID: 8062247, 16395668, 19267393). This variant has been reported as a recurrent de novo mutation in individuals affected with Lynch syndrome-associated cancer in different ethnicities (PMID: 10978353). This variant has been reported in many individuals and families with Lynch syndrome or suspected Lynch syndrome worldwide (PMID: 8062247, 10413423, 10446963, 11920650, 12112654, 12362047, 15222003, 16203774, 16395668, 17312306, 18625694, 19130300, 19419416, 20682701, 21681552, 22883484), and shown to segregate with Lynch syndrome cancers in family studies (PMID: 19267393). This variant is considered to be a founder mutation in the Newfoundland population (PMID: 20682701). This variant has been identified in 1/30582 chromosomes in the general population by the Genome Aggregation Database (gnomAD). Based on the available evidence, this variant is classified as Pathogenic.
PP4, PS2, PVS1
"This variant has been reported in ClinVar as Pathogenic (27 clinical laboratories) and as Uncertain significance (1 clinical laboratories) and as Pathogenic by International Society for Gastrointestinal Hereditary Tumours (InSiGHT) expert panel."
COSMIC Somatic Evidence
Open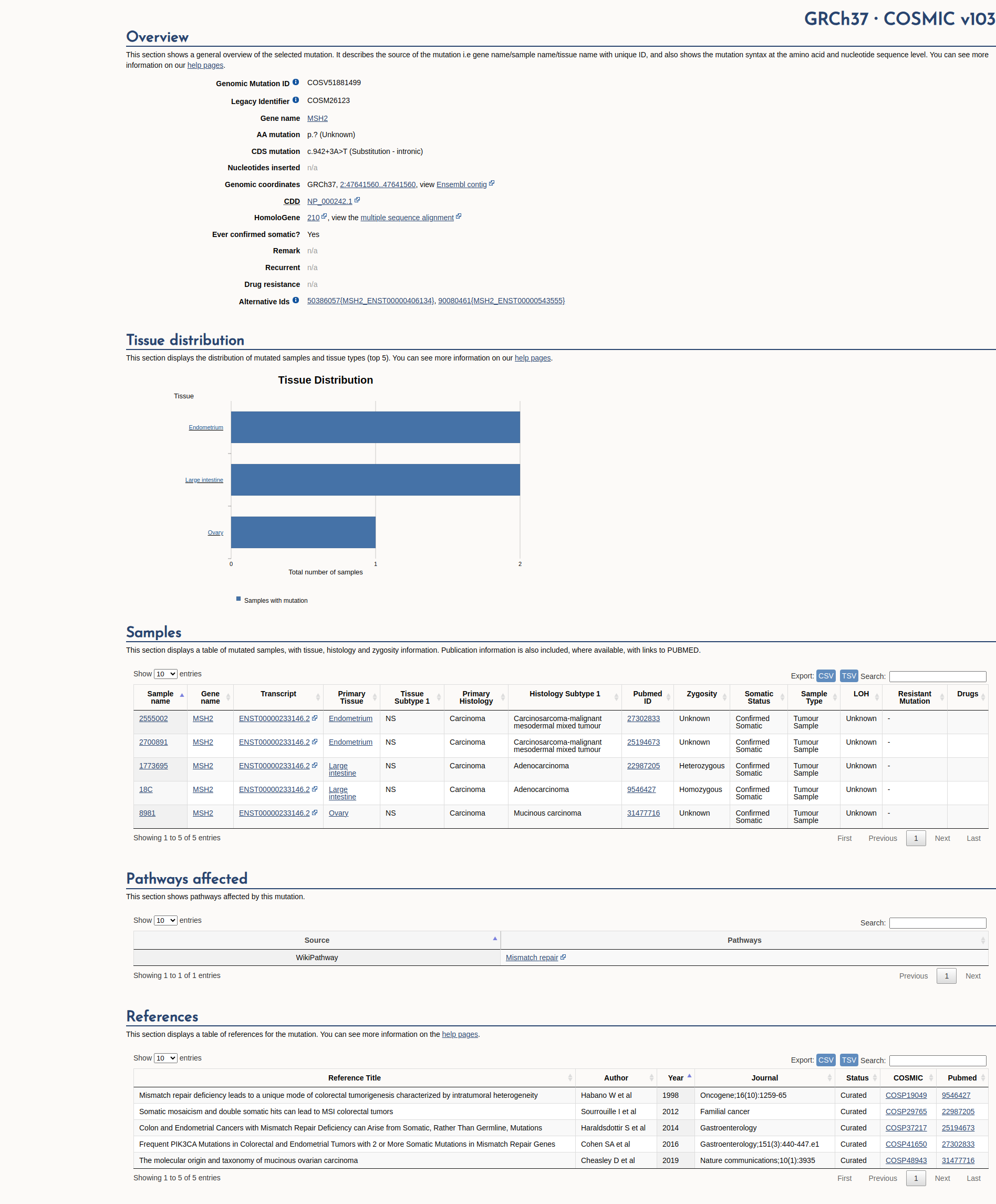
Functional Impact & Domains
Functional Domain
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.42 | -152 bp |
| Donor Loss (DL) | 0.49 | -3 bp |
| Acceptor Gain (AG) | 0.0 | -487 bp |
| Donor Gain (DG) | 0.0 | -129 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Not Applied)
According to VCEP guidelines, the rule for PVS1 is: "Variants at IVS±1 or IVS±2 where exon skipping or use of a cryptic splice site disrupts the reading frame and is predicted to undergo NMD." The evidence for this variant shows it is at position +3 (c.942+3A>T), outside the ±1/2 canonical splice sites. Therefore, this criterion is not applied.
PS1 (Not Applied)
According to VCEP guidelines, PS1 applies to missense substitutions encoding the same amino acid change as a known pathogenic variant. The evidence shows this is an intronic +3 variant with no defined amino acid change. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, PS2 applies to confirmed de novo variants with parental testing. Parental data are not available for this case. Therefore, this criterion is not applied.
PS3 (Not Applied)
According to VCEP guidelines, PS3 requires calibrated functional assays demonstrating loss of MMR function. No functional assay data are available for this variant. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, PS4 requires statistically significant case–control data or multiple affected individuals. No case/control or case series data are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, PM1 applies to variants located in mutational hot spots or well-established functional domains. This variant is intronic outside known domains. Therefore, this criterion is not applied.
PM2 (Not Applied)
According to VCEP guidelines, the rule for PM2 is: "Absent/extremely rare (<1 in 50,000 alleles) in gnomAD v4." The evidence shows a MAF of 0.00327% (~1 in 30,582 alleles), which is above the 1/50,000 threshold. Therefore, this criterion is not applied.
PM3 (Not Applied)
According to standard ACMG guidelines, PM3 applies to recessive diseases with trans observations. Lynch syndrome is dominant and no trans observations are reported. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, PM4 applies to protein length changes (in-frame indels). This intronic variant does not alter protein length directly. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, PM5 applies to novel missense changes at residues with known pathogenic missense variants. This is not a missense variant. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, PM6 applies to presumed de novo variants without confirmation. No parental data exist. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, PP1 requires co-segregation with disease in families. No segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, PP2 applies to missense variants in genes with low tolerance for benign missense change. This variant is intronic. Therefore, this criterion is not applied.
PP3 (Supporting)
According to VCEP guidelines, the rule for PP3 is: "Predicted splice defect for non-canonical splicing nucleotides using SpliceAI with delta score ≥ 0.2." The evidence shows a SpliceAI donor loss delta score of 0.49. Therefore, this criterion is applied at Supporting strength.
PP4 (Not Applied)
According to VCEP guidelines, PP4 requires tumor data (MSI-H or loss of MMR protein) in multiple individuals. No tumor or pathology data are provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, PP5 applies to reputable source assertions without available evidence. ClinGen recommends against using PP5 when expert-panel classifications exist. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to VCEP guidelines, the rule for BA1 is: "Filtering allele frequency ≥ 0.001 (0.1%) in gnomAD v4." The evidence shows a MAF of 0.00327% (<0.1%). Therefore, this criterion is not applied.
BS1 (Not Applied)
According to VCEP guidelines, the rule for BS1 is: "Filtering allele frequency ≥ 0.0001 and < 0.001 (0.01-0.1%) in gnomAD v4." The evidence shows a MAF of 0.00327% (>0.0001 but >0.001?). Actually 0.00327% is 0.0000327, below 0.0001 threshold. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to VCEP guidelines, BS2 requires trans co-occurrence with a pathogenic variant in a patient without CMMRD. No co-occurrence data are available. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to VCEP guidelines, BS3 requires well-validated functional assays showing normal function. No functional data are available. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, BS4 requires lack of segregation in families. No segregation data are available. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, BP1 applies to missense variants in genes where truncating variants are predominant. This variant is intronic. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, BP2 applies to observed in cis with pathogenic variant for dominant. No such data exist. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, BP3 applies to in-frame repeats. Not relevant to this intronic variant. Therefore, this criterion is not applied.
BP4 (Not Applied)
According to VCEP guidelines, BP4 for intronic variants requires SpliceAI delta score ≤ 0.1. The evidence shows delta = 0.49. Therefore, this criterion is not applied.
BP5 (Not Applied)
According to standard ACMG guidelines, BP5 applies to variant found in a case with an alternate cause. No such tumor or alternate cause data are available. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, BP6 applies to unpublished, unverified assertions of benign impact. No such assertions exist. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to VCEP guidelines, the rule for BP7 is: "Synonymous or intronic variants at or beyond -21/+7 with no splice impact." This variant is at +3. Therefore, this criterion is not applied.