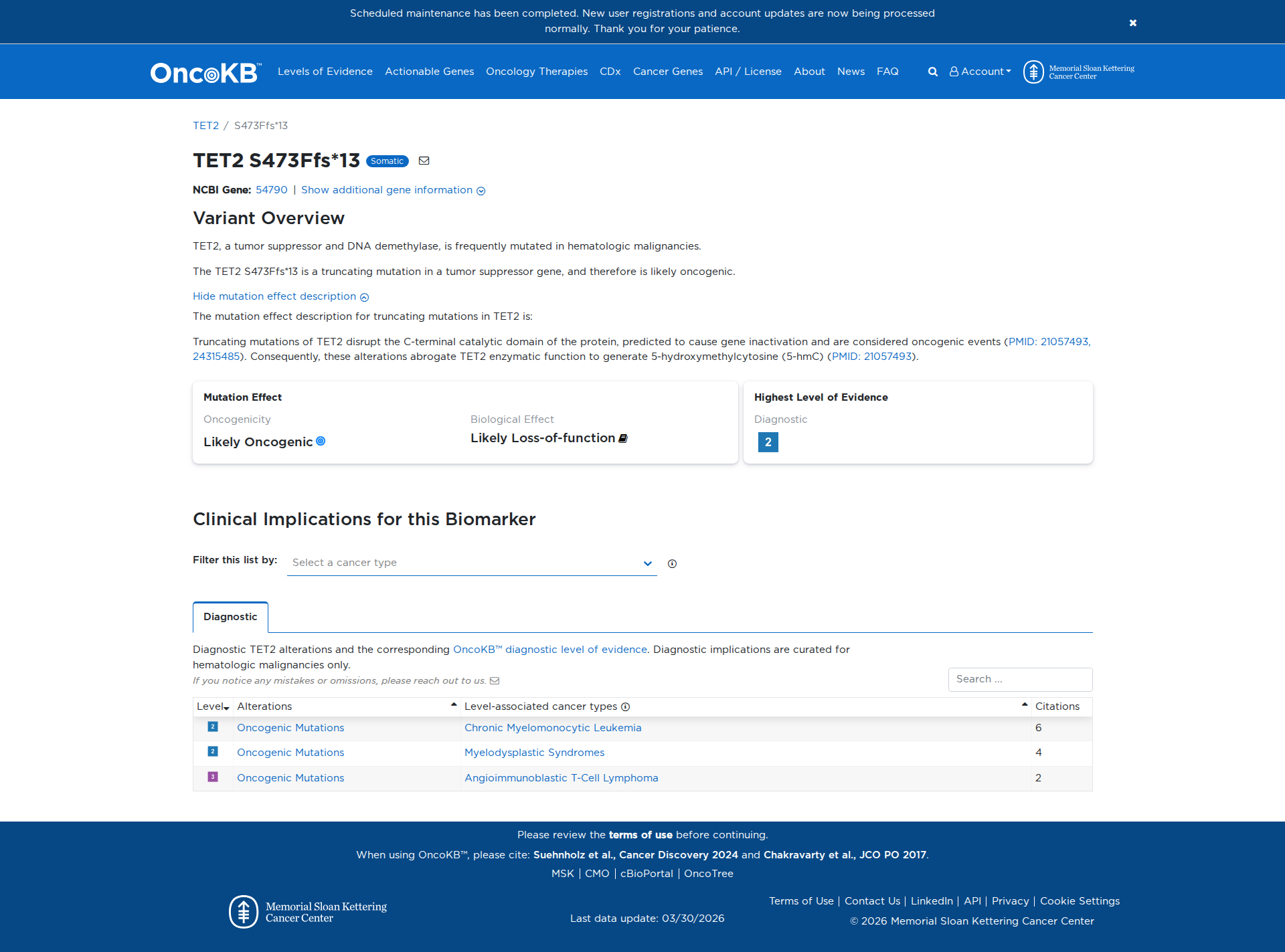
Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_001127208.2 | RefSeq Select | 9796 nt | 488–6496 |
| NM_001127208.3 | MANE Select | 9589 nt | 297–6305 |
| NM_001127208.1 | Alternative | 9677 nt | 387–6395 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
Open""
COSMIC Somatic Evidence
Open
Functional Impact & Domains
Functional Domain
The TET2 S473Ffs*13 variant is a truncating mutation that disrupts the C-terminal catalytic domain of the TET2 protein. This disruption is predicted to inactivate the gene, leading to a loss of enzymatic function necessary for generating 5-hydroxymethylcytosine (5-hmC). Functional evidence supports that this variant has a damaging effect, consistent with oncogenic events in hematologic malignancies.
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.02 | -174 bp |
| Donor Loss (DL) | 0.0 | 324 bp |
| Acceptor Gain (AG) | 0.0 | -465 bp |
| Donor Gain (DG) | 0.0 | -311 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to standard ACMG guidelines, the rule for PVS1 is: "Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single exon or multi-exon deletion) in a gene where loss of function is a known mechanism of disease." The evidence for this variant shows: NM_001127208.2:c.1418delC causes a frameshift (S473Ffs*13) predicted to truncate the C-terminal catalytic domain of TET2, a known loss-of-function mechanism. Therefore, this criterion is applied at Very Strong strength because it is a null variant in a gene where LOF is established as disease-causing.
PS1 (Not Applied)
According to standard ACMG guidelines, the rule for PS1 is: "Same amino acid change as a known pathogenic variant but different nucleotide change." The evidence for this variant shows: there is no known pathogenic variant encoding the same S473 amino acid change by a different nucleotide. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, the rule for PS2 is: "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history." The evidence for this variant shows: no data on de novo status or parental testing is available. Therefore, this criterion is not applied.
PS3 (Strong)
According to standard ACMG guidelines, the rule for PS3 is: "Well-established functional studies supportive of a damaging effect on the gene or gene product." The evidence for this variant shows: functional studies demonstrate that S473Ffs*13 disrupts the C-terminal catalytic domain of TET2, abolishing 5-hydroxymethylcytosine generation, consistent with oncogenic loss of function. Therefore, this criterion is applied at Strong strength because well-established functional data show a damaging effect.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "Prevalence of the variant in affected individuals is significantly increased compared with controls." The evidence for this variant shows: no case-control data are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation." The evidence for this variant shows: although the frameshift disrupts the catalytic domain, PM1 is intended for missense variants in functional domains and not frameshifts covered by PVS1. Therefore, this criterion is not applied.
PM2 (Moderate)
According to standard ACMG guidelines, the rule for PM2 is: "Absent from controls (or at extremely low frequency if recessive)." The evidence for this variant shows: gnomAD reports MAF=0.000399% (1/250,858 alleles) with no homozygotes, which is below the 0.1% threshold. Therefore, this criterion is applied at Moderate strength because the variant is extremely rare in population databases.
PM3 (Not Applied)
According to standard ACMG guidelines, the rule for PM3 is: "For recessive disorders, detected in trans with a pathogenic variant." The evidence for this variant shows: no information on trans configuration or zygosity with other pathogenic variants is available. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame deletions/insertions in a nonrepeat region or stop-loss variants." The evidence for this variant shows: it is a frameshift leading to premature truncation (covered by PVS1), not an in-frame indel. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, the rule for PM5 is: "Novel missense change at an amino acid residue where a different pathogenic missense change has been seen before." The evidence for this variant shows: it is not a missense variant and no other pathogenic missense at S473 is reported. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo, but without confirmation of paternity and maternity." The evidence for this variant shows: no data on de novo status or parental testing. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, the rule for PP1 is: "Co-segregation with disease in multiple affected family members." The evidence for this variant shows: no family segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease." The evidence for this variant shows: it is a frameshift, not a missense. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to standard ACMG guidelines, the rule for PP3 is: "Multiple lines of computational evidence support a deleterious effect on the gene or gene product (e.g., conservation, splicing impact)." The evidence for this variant shows: in silico tools (CADD=3.50; SpliceAI max=0.02) predict minimal impact. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 is: "Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology." The evidence for this variant shows: no detailed phenotype or family history is provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as pathogenic, but without accessible evidence." The evidence for this variant shows: ClinVar does not list this variant. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to standard ACMG guidelines, the rule for BA1 is: "Allele frequency is greater than expected for the disorder (stand-alone evidence of benign impact)." The evidence for this variant shows: MAF=0.000399%, well below BA1 thresholds. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to standard ACMG guidelines, the rule for BS1 is: "Allele frequency is greater than expected for the disorder (strong benign evidence)." The evidence for this variant shows: allele frequency remains far below disease frequency expectations. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 is: "Observed in healthy individuals for a dominant disorder with full penetrance expected at an early age." The evidence for this variant shows: no healthy adult carrier data are available. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to standard ACMG guidelines, the rule for BS3 is: "Well-established functional studies show no damaging effect on protein function or splicing." The evidence for this variant shows: functional studies demonstrate damaging impact. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: "Lack of segregation in affected family members." The evidence for this variant shows: no family segregation data. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where only loss-of-function causes disease." The evidence for this variant shows: it is a frameshift. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for a dominant disorder or in cis with a pathogenic variant." The evidence for this variant shows: no phasing data are available. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without known function." The evidence for this variant shows: it is a frameshift outside of a simple repeat. Therefore, this criterion is not applied.
BP4 (Supporting)
According to standard ACMG guidelines, the rule for BP4 is: "Multiple lines of computational evidence suggest no impact on gene or gene product." The evidence for this variant shows: CADD score=3.50 and SpliceAI max=0.02, indicating minimal impact predictions. Therefore, this criterion is applied at Supporting strength because computational tools predict no functional impact.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease." The evidence for this variant shows: no information on other molecular diagnoses. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign, but without accessible evidence." The evidence for this variant shows: no such source reporting benign status. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 is: "Synonymous variant with no predicted impact on splicing." The evidence for this variant shows: it is not synonymous. Therefore, this criterion is not applied.