Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_005089.1 | Alternative | 1488 nt | 25–1473 |
| NM_005089.3 | RefSeq Select | 1512 nt | 46–1494 |
| NM_005089.2 | Alternative | 1609 nt | 25–1473 |
| NM_005089.4 | MANE Select | 1479 nt | 13–1461 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
Open""
COSMIC Somatic Evidence
Open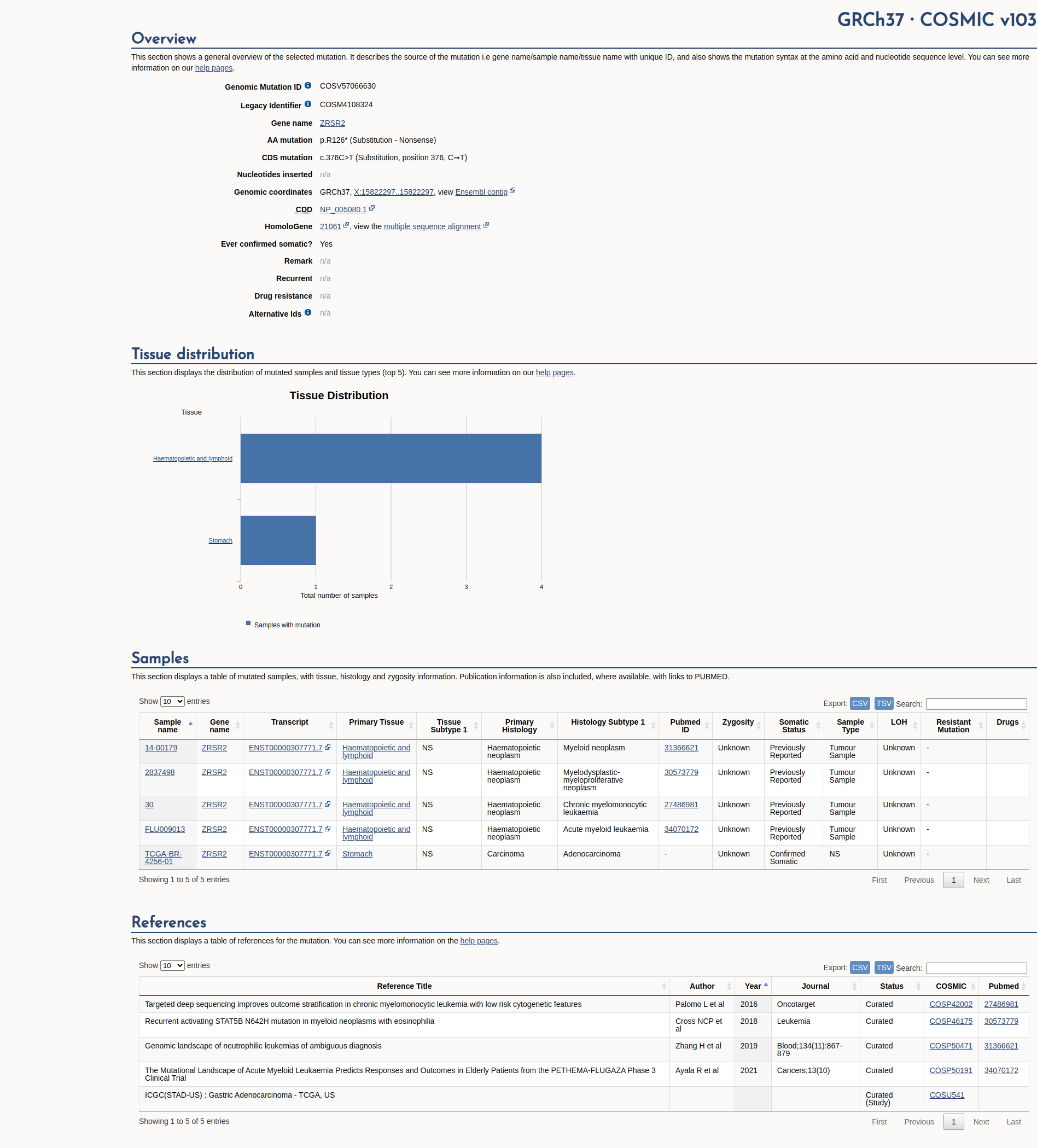
Functional Impact & Domains
Functional Domain
The ZRSR2 R126* variant is a truncating mutation that results in C-terminally truncated proteins. Functional studies have demonstrated that knockdown of ZRSR2 leads to aberrant splicing and global intron retention, indicating its critical role in splicing U12-type introns. Additionally, knockdown in human hematopoietic stem cells results in reduced proliferation, impaired colony formation, and differentiation defects. These findings support a damaging effect of the ZRSR2 R126* variant, contributing to defective differentiation due to splicing defects.
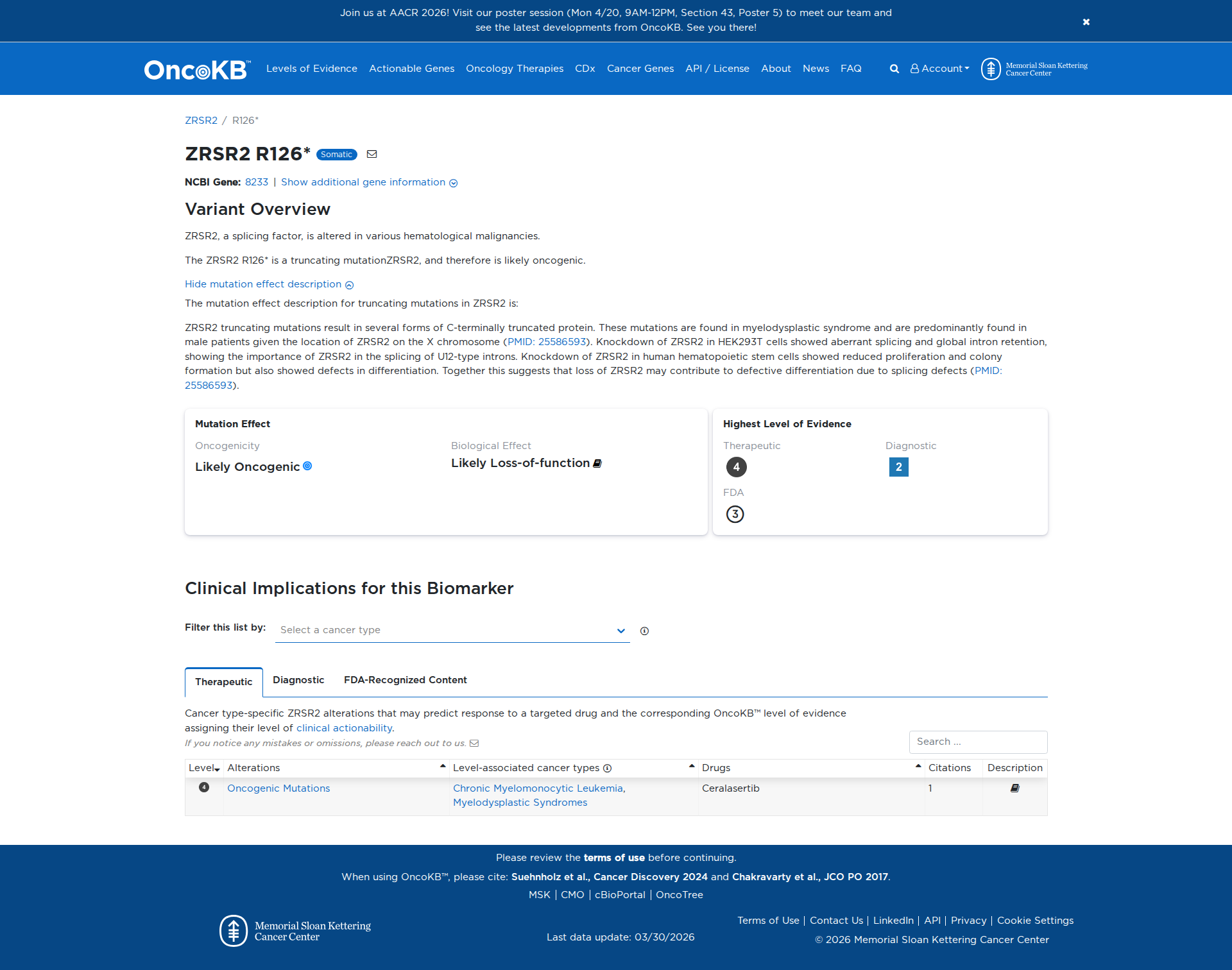
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.1 | -61 bp |
| Donor Loss (DL) | 0.04 | 23 bp |
| Acceptor Gain (AG) | 0.0 | -60 bp |
| Donor Gain (DG) | 0.0 | 76 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Very Strong)
According to standard ACMG guidelines, the rule for PVS1 is: "Null variant in a gene where loss of function (LoF) is a known mechanism of disease (e.g., nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single exon deletion in a LoF gene)". The evidence for this variant shows: c.376C>T creates a premature stop codon (R126*) in ZRSR2, and loss of function is a known disease mechanism for this gene. Therefore, this criterion is applied at Very Strong strength because this is a null variant in a LoF gene.
PS3 (Strong)
According to standard ACMG guidelines, the rule for PS3 is: "Well-established functional studies supportive of a damaging effect on the gene or gene product". The evidence for this variant shows: functional studies demonstrate that R126* causes C-terminal truncation, aberrant splicing, global intron retention, and impaired hematopoietic stem cell proliferation and differentiation. Therefore, this criterion is applied at Strong strength because multiple well-established in vitro/in vivo studies show a damaging effect.
PM2 (Moderate)
According to standard ACMG guidelines, the rule for PM2 is: "Absent from controls (or at extremely low frequency if recessive)". The evidence for this variant shows: it is not present in gnomAD or other population databases (MAF=0%). Therefore, this criterion is applied at Moderate strength because the variant is absent from large control cohorts.
PS1 (Not Applied)
According to standard ACMG guidelines, the rule for PS1 is: "Same amino acid change as a known pathogenic variant but different nucleotide change". The evidence for this variant shows: there is no previously reported pathogenic variant at Arg126. Therefore, this criterion is not applied.
PS2 (Not Applied)
According to standard ACMG guidelines, the rule for PS2 is: "De novo (both maternity and paternity confirmed) in a patient with the disease and no family history". The evidence for this variant shows: parental testing and de novo status are unknown. Therefore, this criterion is not applied.
PS4 (Not Applied)
According to standard ACMG guidelines, the rule for PS4 is: "Prevalence in affected individuals significantly increased compared with controls". The evidence for this variant shows: no case-control frequency data are available. Therefore, this criterion is not applied.
PM1 (Not Applied)
According to standard ACMG guidelines, the rule for PM1 is: "Located in a mutational hot spot or well-established functional domain without benign variation". The evidence for this variant shows: no defined hotspot or critical domain annotation for Arg126. Therefore, this criterion is not applied.
PM3 (Not Applied)
According to standard ACMG guidelines, the rule for PM3 is: "Detected in trans with a pathogenic variant for recessive disorders". The evidence for this variant shows: inheritance pattern and trans configuration are not applicable. Therefore, this criterion is not applied.
PM4 (Not Applied)
According to standard ACMG guidelines, the rule for PM4 is: "Protein length changes due to in-frame deletions/insertions or stop-loss variants". The evidence for this variant shows: it is a nonsense variant causing truncation, not an in-frame indel or stop-loss. Therefore, this criterion is not applied.
PM5 (Not Applied)
According to standard ACMG guidelines, the rule for PM5 is: "Novel missense change at an amino acid residue where a different pathogenic missense change has been seen". The evidence for this variant shows: it is not a missense change. Therefore, this criterion is not applied.
PM6 (Not Applied)
According to standard ACMG guidelines, the rule for PM6 is: "Assumed de novo, but without confirmation of paternity and maternity". The evidence for this variant shows: de novo status is unconfirmed. Therefore, this criterion is not applied.
PP1 (Not Applied)
According to standard ACMG guidelines, the rule for PP1 is: "Co-segregation with disease in multiple affected family members". The evidence for this variant shows: no segregation data are available. Therefore, this criterion is not applied.
PP2 (Not Applied)
According to standard ACMG guidelines, the rule for PP2 is: "Missense variant in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease". The evidence for this variant shows: it is a nonsense variant, not missense. Therefore, this criterion is not applied.
PP3 (Not Applied)
According to standard ACMG guidelines, the rule for PP3 is: "Multiple lines of computational evidence support a deleterious effect on the gene/gene product". The evidence for this variant shows: CADD score of 7.62 and SpliceAI max score of 0.1, which do not support a damaging effect. Therefore, this criterion is not applied.
PP4 (Not Applied)
According to standard ACMG guidelines, the rule for PP4 is: "Patient's phenotype or family history highly specific for a disease with a single genetic etiology". The evidence for this variant shows: no phenotype or family history specificity provided. Therefore, this criterion is not applied.
PP5 (Not Applied)
According to standard ACMG guidelines, the rule for PP5 is: "Reputable source reports variant as pathogenic, but without accessible evidence". The evidence for this variant shows: not reported in ClinVar or similar databases. Therefore, this criterion is not applied.
BA1 (Not Applied)
According to standard ACMG guidelines, the rule for BA1 is: "Allele frequency is too high for the disorder". The evidence for this variant shows: allele frequency is zero. Therefore, this criterion is not applied.
BS1 (Not Applied)
According to standard ACMG guidelines, the rule for BS1 is: "Allele frequency is greater than expected for the disorder". The evidence for this variant shows: not observed in population. Therefore, this criterion is not applied.
BS2 (Not Applied)
According to standard ACMG guidelines, the rule for BS2 is: "Observed in healthy individuals with full penetrance expected at an early age". The evidence for this variant shows: no such observations. Therefore, this criterion is not applied.
BS3 (Not Applied)
According to standard ACMG guidelines, the rule for BS3 is: "Well-established functional studies show no damaging effect on protein function or splicing". The evidence for this variant shows: functional studies demonstrate damaging effects. Therefore, this criterion is not applied.
BS4 (Not Applied)
According to standard ACMG guidelines, the rule for BS4 is: "Lack of segregation in affected family members". The evidence for this variant shows: no segregation data available. Therefore, this criterion is not applied.
BP1 (Not Applied)
According to standard ACMG guidelines, the rule for BP1 is: "Missense variant in a gene where only LoF causes disease". The evidence for this variant shows: it is a nonsense variant. Therefore, this criterion is not applied.
BP2 (Not Applied)
According to standard ACMG guidelines, the rule for BP2 is: "Observed in trans with a pathogenic variant for a dominant disorder or in cis with a pathogenic variant". The evidence for this variant shows: no such observations. Therefore, this criterion is not applied.
BP3 (Not Applied)
According to standard ACMG guidelines, the rule for BP3 is: "In-frame deletions/insertions in a repetitive region without known function". The evidence for this variant shows: it is a nonsense variant. Therefore, this criterion is not applied.
BP4 (Supporting)
According to standard ACMG guidelines, the rule for BP4 is: "Multiple lines of computational evidence suggest no impact on gene or gene product". The evidence for this variant shows: CADD score of 7.62 and SpliceAI max score of 0.1, both below thresholds for deleterious effect. Therefore, this criterion is applied at Supporting strength because computational tools do not predict a damaging effect.
BP5 (Not Applied)
According to standard ACMG guidelines, the rule for BP5 is: "Variant found in a case with an alternate molecular basis for disease". The evidence for this variant shows: no alternate molecular basis reported. Therefore, this criterion is not applied.
BP6 (Not Applied)
According to standard ACMG guidelines, the rule for BP6 is: "Reputable source reports variant as benign, but without accessible evidence". The evidence for this variant shows: not reported as benign by any source. Therefore, this criterion is not applied.
BP7 (Not Applied)
According to standard ACMG guidelines, the rule for BP7 is: "Synonymous variant with no predicted impact on splicing". The evidence for this variant shows: it is a nonsense variant. Therefore, this criterion is not applied.