Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_001369786.1 | Alternative | 5417 nt | 178–747 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.65A>G variant in the KRAS gene is a missense variant predicted to cause substitution of glutamine by arginine at amino acid 22 (p.Glln22Arg). This variant is absent from gnomAD v2.1.1 (PM2_Supporting). The computational predictor REVEL gives a score of 0.749 supporting a deleterious impact to KRAS function (PP3). The variant is located in the KRAS gene, which has been defined by the ClinGen RASopathy Expert Panel as a gene with a low rate of benign missense variants and pathogenic missense variants are common (PP2). This variant has been reported in several individuals with clinical features of a RASopathy, with 2 unconfirmed de novo occurrences (PS4, PM6_VeryStrong; GeneDx, Partners LMM, HudsonAlpha Institute for Biotechnology, Center for Genomics, Ann and Robert H. Lurie Children's Hospital of Chicago, ARUP Laboratories, Molecular Genetics and Genomics; ClinVar: SCV000207881.10; SCV000198473.4; SCV001870321.1; SCV000678231.1; SCV000604082.1). In vitro functional studies showed that this variant enhanced ERK activation (PS3_Supporting; PMID: 20949621). In summary, this variant meets criteria to be classified as pathogenic for autosomal dominant RASopathies based on the ACMG/AMP criteria applied, as specified by the ClinGen RASopathy Variant Curation Expert Panel: PM6_VeryStrong, PS4, PS3_Supporting, PM2_Supporting, PP2, PP3 (Specification Version 2.3, 12/3/2024)
The p.Gln22Arg variant in KRAS has been reported in 8 individuals with clinical features of a RASopathy, and occured de novo in 1 of these individuals (Zenker 2007, Gremer 2010, LMM unpublished data). It was absent from large population st udies. In vitro functional studies provide some evidence that the p.Gln22Arg var iant impacts protein function (Gremer 2010). In summary, this variant meets our criteria to be classified as pathogenic for Noonan syndrome in an autosomal domi nant manner based upon de novo occurrence, absence from controls, and functional evidence.
This sequence change replaces glutamine, which is neutral and polar, with arginine, which is basic and polar, at codon 22 of the KRAS protein (p.Gln22Arg). This variant is not present in population databases (gnomAD no frequency). This missense change has been observed in individuals with Noonan syndrome (PMID: 17056636, 29948256; internal data). ClinVar contains an entry for this variant (Variation ID: 40452). Invitae Evidence Modeling incorporating data from in vitro experimental studies (internal data) indicates that this missense variant is expected to disrupt KRAS function with a positive predictive value of 95%. Experimental studies have shown that this missense change affects KRAS function (PMID: 20949621). This variant disrupts the p.Glu22 amino acid residue in KRAS. Other variant(s) that disrupt this residue have been determined to be pathogenic (PMID: 17056636, 17324647, 20949621, 24803665). This suggests that this residue is clinically significant, and that variants that disrupt this residue are likely to be disease-causing. For these reasons, this variant has been classified as Pathogenic.
Same nucleotide change resulting in same amino acid change has been previously reported as pathogenic/likely pathogenic with strong evidence (ClinVar ID: VCV000040452). Functional studies provide strong evidence of the variant having a damaging effect on the gene or gene product (PMID: 20652921). A different missense change at the same codon has been reported as pathogenic/likely pathogenic with strong evidence (ClinVar ID: VCV000040451,VCV000040453,VCV000376325, PMID:17056636). In silico tool predictions suggest damaging effect of the variant on gene or gene product (REVEL: 0.749>=0.6). A missense variant is a common mechanism associated with Noonan syndrome 3. It is not observed in the gnomAD v2.1.1 dataset. Therefore, this variant is classified as pathogenic according to the recommendation of ACMG/AMP guideline.
"This variant has been reported in ClinVar as Pathogenic (12 clinical laboratories) and as Pathogenic by ClinGen RASopathy Variant Curation Expert Panel expert panel."
COSMIC Somatic Evidence
Open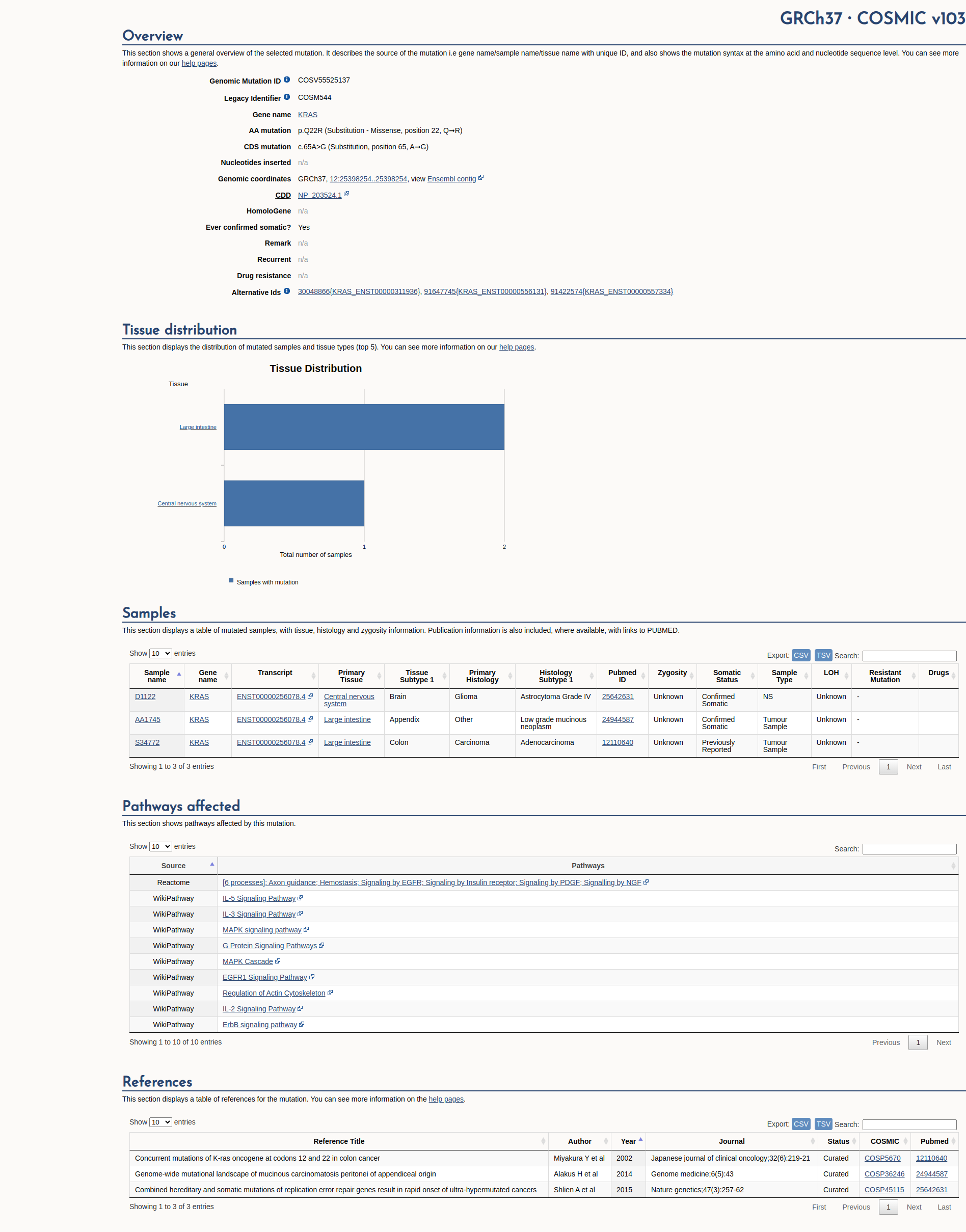
Functional Impact & Domains
Functional Domain
Error in OpenAI Consolidation. OncoKB: KRASQ22RKRASQ22RSomaticNCBI Gene:3845|Show additional gene information Variant OverviewKRAS, a GTPase which functions as an upstream regulator of the MAPK pathway, is frequently mutated in various cancer types including lung, colorectal and pancreatic cancers.The KRAS Q22R mutation is likely oncogenic.Hide mutation effect description The KRAS Q22R mutation is located in the catalytic G-domain of the protein. This mutation was found as a germline mutation in Noonan syndrome (PMID: 21263000). Expression of this mutation in vitro and in a simian cell line demonstrated that it is activating as measured by increased protein and pathway activation compared to wildtype (PMID: 20949621). JAX-CKB: No results found
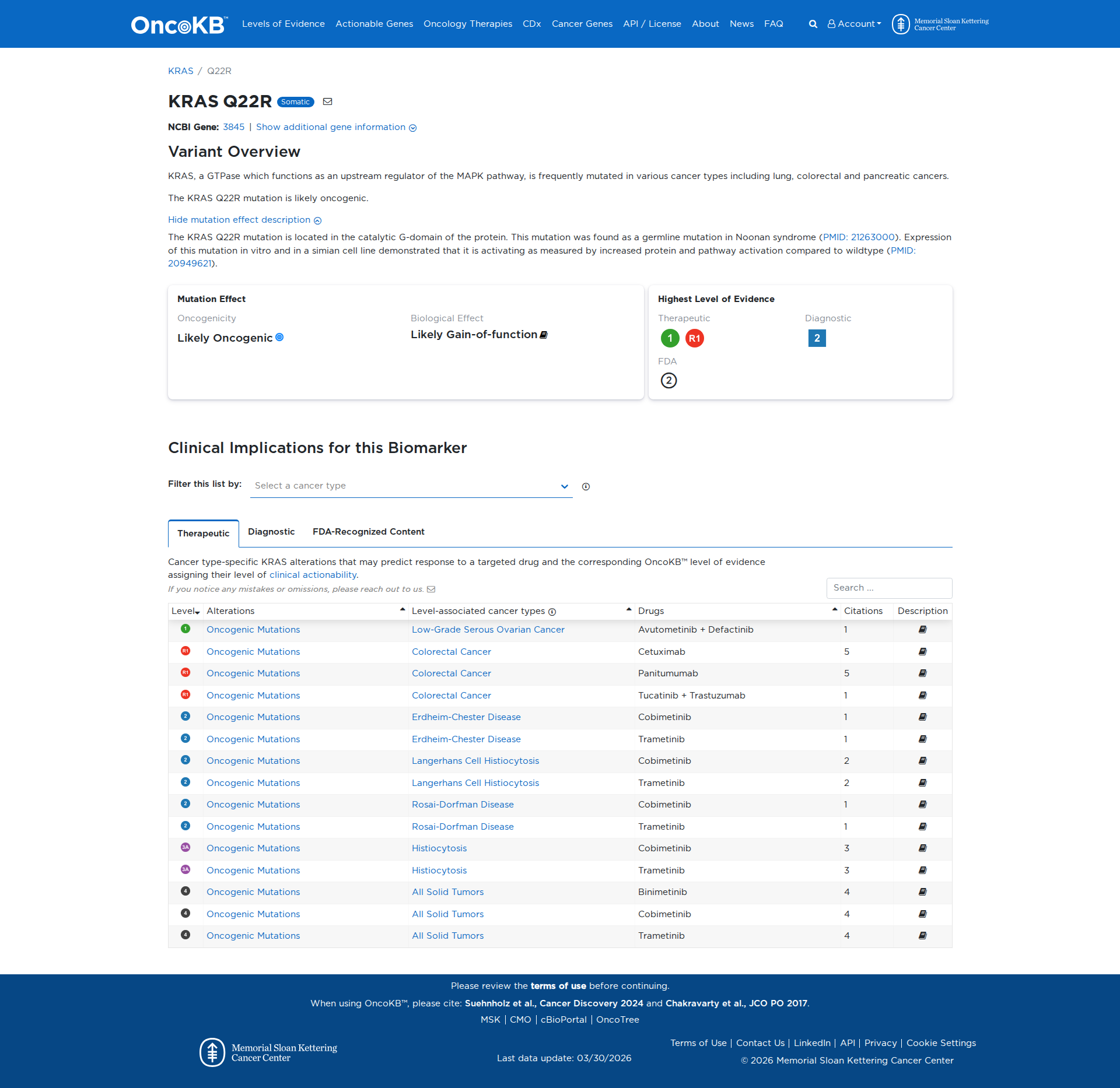
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | None bp |
| Donor Loss (DL) | 0.0 | None bp |
| Acceptor Gain (AG) | 0.0 | None bp |
| Donor Gain (DG) | 0.0 | None bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PS3 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PM2 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PP5 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)