Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_006218.2 | Alternative | 3724 nt | 158–3364 |
| NM_006218.3 | Alternative | 9104 nt | 158–3364 |
| NM_006218.4 | MANE Select | 9259 nt | 324–3530 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.2176G>A (NM_006218.4) variant in PIK3CA is a missense variant predicted to cause substitution of (p.Glu726Lys). This variant is absent from gnomAD v2.1.1 (PM2_Supporting). PIK3CA, in which the variant was identified, is defined by the ClinGen Brain Malformations Expert Panel as a gene that has a low rate of benign missense variation and where pathogenic missense variants are a common mechanism of disease (PP2). The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls (PS4_VS; identified in at least 15 individuals with a clinical diagnosis of megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome; (MPPH) or megalencephaly-capillary malformation-polymicrogyria syndrome; (MCAP), it has been shown to significantly increase phosphorylation levels in patient cell lines (PMID: 28566443), and is in at least 15 tumor samples in the literature and COSMIC (PMID: 22729224, PMID: 28941273, PMID: 24497998 )). This variant has been confirmed de novo and has been identified with variable allelic fractions consistent with a post-zygotic event (PS2_Strong; PMIDs: 22729224, 22729224). In summary, this variant meets the criteria to be classified as Pathogenic for mosaic autosomal dominant overgrowth with or without cerebral malformations due to abnormalities in MTOR-pathway genes based on the ACMG/AMP criteria applied, as specified by the ClinGen Brain Malformations Expert Panel: PM2_P, PP2, PS4_VS, PS2; 14 points (VCEP specifications version 1; Approved: 1/31/2021)
The variant is not observed in the gnomAD v2.1.1 dataset. Missense changes are a common disease-causing mechanism. Same nucleotide change resulting in same amino acid change has been previously reported as pathogenic/likely pathogenic with strong evidence (ClinVar ID: VCV000376476). The variant has been previously reported as de novo in a similarly affected individual (PMID: 27631024). The variant has been observed in at least two similarly affected unrelated individuals (PMID: 22729224). Different missense changes at the same codon (p.Glu726Ala, p.Glu726Gly) have been reported to be associated with PIK3CA-related disorder (ClinVar ID: VCV000376477 / PMID: 29549527). Therefore, this variant is classified as Pathogenic according to the recommendation of ACMG/AMP guideline.
The c.2176G>A (p.E726K) alteration is located in exon 14 (coding exon 13) of the PIK3CA gene. This alteration results from a G to A substitution at nucleotide position 2176, causing the glutamic acid (E) at amino acid position 726 to be replaced by a lysine (K). This variant was not reported in population-based cohorts in the Genome Aggregation Database (gnomAD). This variant has been determined to be the result of a de novo post-zygotic mutation in multiple individual with features consistent with PIK3CA-related disorders (Rivière, 2012; Tapper, 2014; McDermott, 2016; Mirzaa, 2016; Kuentz, 2017; McDermott, 2017; Ambry internal data). This amino acid position is highly conserved in available vertebrate species. This missense alteration is located in a region that has a low rate of benign missense variation (Lek, 2016; Firth, 2009). A functional analysis of patient derived cells have shown a significant increase of phosphorylation levels for p.E726K (Leiter, 2017). This alteration is predicted to be deleterious by in silico analysis. Based on the available evidence, this alteration is classified as pathogenic.
This sequence change replaces glutamic acid, which is acidic and polar, with lysine, which is basic and polar, at codon 726 of the PIK3CA protein (p.Glu726Lys). This variant is not present in population databases (gnomAD no frequency). This missense change has been observed in individual(s) with megalencephaly-capillary malformation syndrome and/or PIK3CA-related conditions (PMID: 22729224, 24497998, 26351730, 27631024; internal data). In at least one individual the variant was observed to be de novo. ClinVar contains an entry for this variant (Variation ID: 376476). Invitae Evidence Modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) has been performed for this missense variant. However, the output from this modeling did not meet the statistical confidence thresholds required to predict the impact of this variant on PIK3CA protein function. For these reasons, this variant has been classified as Pathogenic.
"This variant has been reported in ClinVar as Pathogenic (7 clinical laboratories) and as Pathogenic by ClinGen Brain Malformations Variant Curation Expert Panel expert panel."
COSMIC Somatic Evidence
Open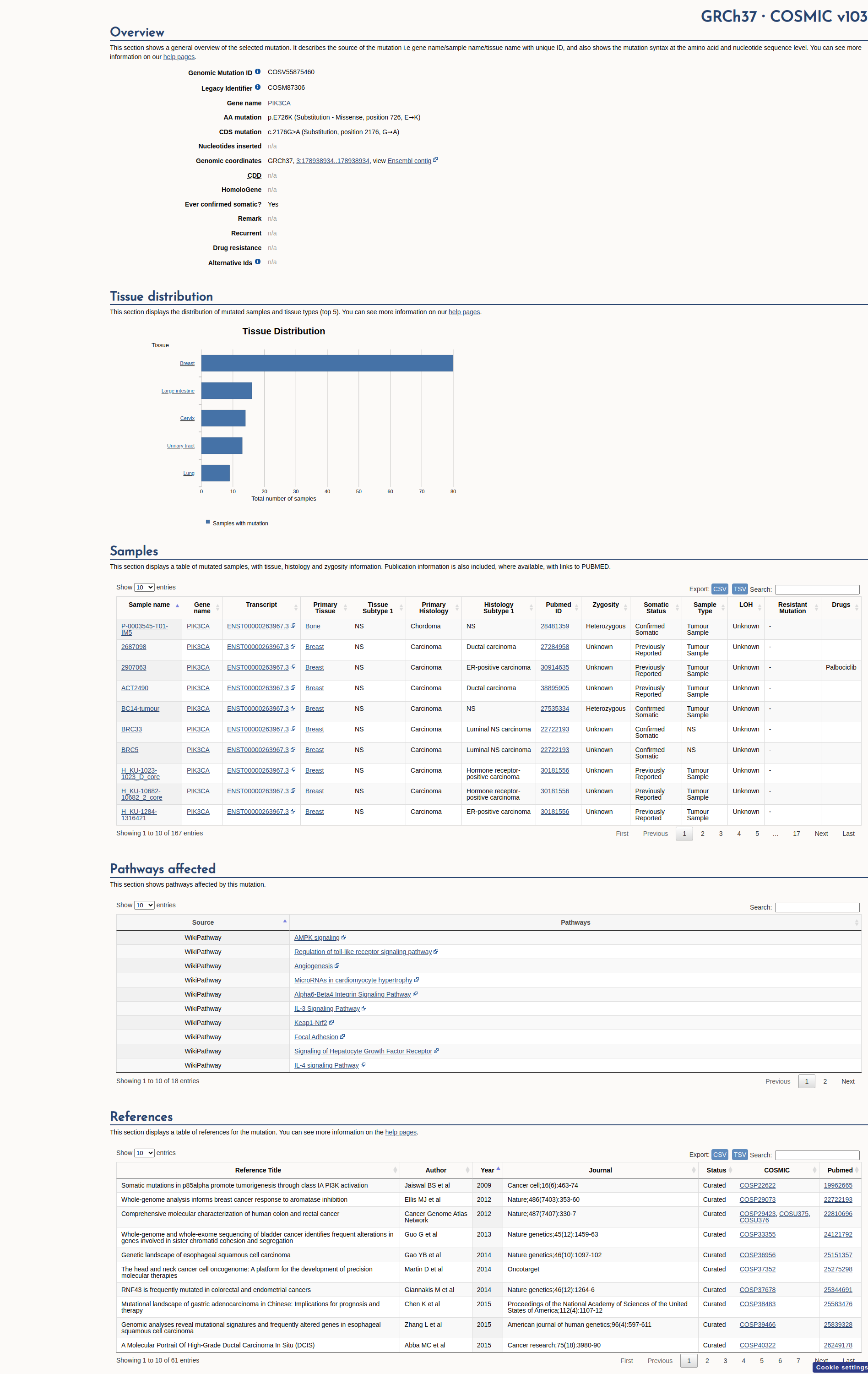
Functional Impact & Domains
Functional Domain
Error in OpenAI Consolidation. OncoKB: PIK3CAE726KPIK3CAE726KSomaticNCBI Gene:5290|Show additional gene information Variant OverviewPIK3CA, the catalytic subunit of PI3-kinase, is frequently mutated in a diverse range of cancers including breast, endometrial and cervical cancers.There is conflicting and/or weak data describing the biological significance of the PIK3CA E726K mutation.Hide mutation effect description The PIK3CA E726K mutation is located between the accessory and the kinase domains of the protein. There is conflicting functional data for this mutation. Expression of this mutation in Ba/F3 but not MCF10A cell lines led to proliferation in the absence of growth factors (PMID: 29533785). JAX-CKB: PIK3CA E726K does not lie within any known functional domains of the Pik3ca protein (UniProt.org). E726K results in increased cell proliferation and migration and increased Akt phosphorylation compared to wild-type Pik3ca in culture (PMID: 34779417) and increased transformation ability in several different cell lines in culture (PMID: 29533785, PMID: 34779417).
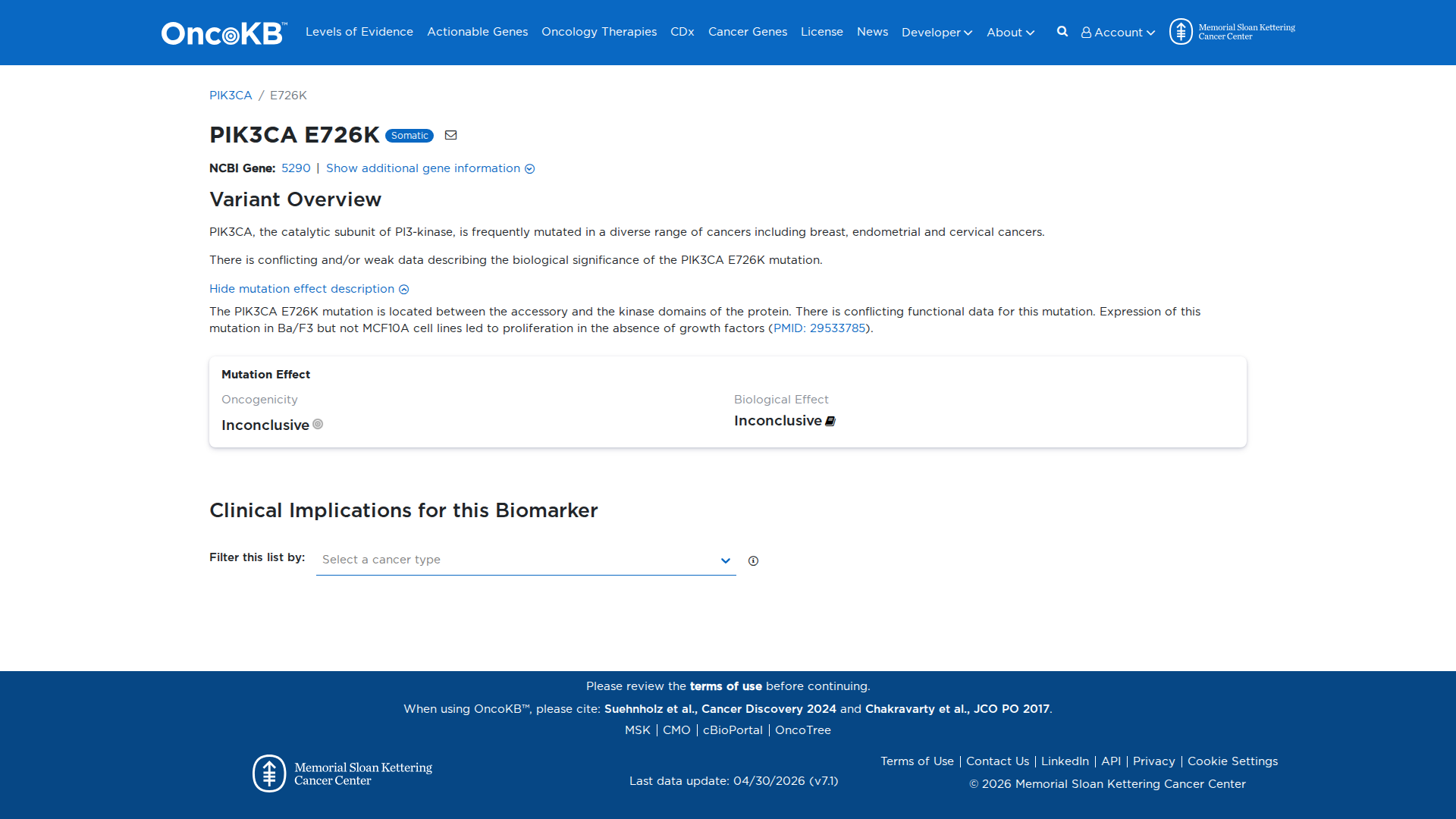

Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.01 | -160 bp |
| Donor Loss (DL) | 0.0 | 11 bp |
| Acceptor Gain (AG) | 0.0 | -301 bp |
| Donor Gain (DG) | 0.02 | -65 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PM2 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PP5 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
BP4 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)