Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_000059.4 | MANE Select | 11954 nt | 200–10456 |
| NM_000059.2 | Alternative | 11386 nt | 228–10484 |
| NM_000059.3 | RefSeq Select | 11386 nt | 228–10484 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenThe c.5946delT (p.S1982Rfs*22) alteration, located in exon 11 (coding exon 10) of the BRCA2 gene, consists of a deletion of one nucleotide at position 5946, causing a translational frameshift with a predicted alternate stop codon after 22 amino acids. This alteration is expected to result in loss of function by premature protein truncation or nonsense-mediated mRNA decay. Based on data from gnomAD, this allele has an overall frequency of 0.028% (78/282088) total alleles studied. The highest observed frequency was 0.589% (61/10364) of Ashkenazi Jewish alleles. This variant is one of the well-described Ashkenazi Jewish founder mutations and has been reported in numerous families affected with breast, ovarian, prostate, pancreatic, and other HBOC-related cancers (Agalliu, 2009; Walsh, 2011; Johnston, 2012; Bayraktar, 2012; George, 2013; Lucas, 2013; Salo-Mullen, 2015; Susswein, 2016). This variant has also been reported in trans with a second variant in individuals affected with Fanconi anemia (Offit, 2003; Dewire, 2009). One study indicated that carriers of the c.5946delT mutation may have a lower relative risk for breast cancer when compared to carriers of other non-Ashkenazi Jewish BRCA2 mutations (Finkelman, 2012); however, other studies have reported the overall risk as similar to that of other pathogenic mutations in the ovarian cancer cluster region (OCCR) of coding exon 10 of BRCA2 (Kuchenbaecker, 2017). This variant is also designated as 6174delT in published literature. Based on the available evidence, this alteration is classified as pathogenic.
This submission and the accompanying classification are no longer maintained by the submitter. For more information on current observations and classification, please contact variantquestions@myriad.com.
The BRCA2 c.5946delT (p.Ser1982ArgfsTer22) variant, more commonly known as c.6174delT, results in a frameshift and premature termination of the protein. The p.Ser1982ArgfsTer22 variant is a well-described founder variant in the BRCA2 gene that is prevalent in the Ashkenazi Jewish and Icelandic populations, with a carrier frequency of ~1.5% (Roa et al. 1996; Neuhausen et al. 1996). The variant has been shown to occur in approximately eight percent of women diagnosed with breast cancer before the age of 42 years (Neuhausen et al. 1996; Oddoux et al. 1996; Petrucelli et al. 2010; Finkelman et al. 2012). By the age of 70, 43% of individuals who carry this variant are predicted to develop breast cancer and 20% are predicted to develop ovarian cancer (Struewing et al. 1997; King et al. 2003). Across a selection of the literature, the p.Ser1982ArgfsTer22 variant has been identified in 55 of 1,272 (4%) individuals with breast cancer, 44 of 382 (11.5%) individuals with ovarian cancer, and at least 118 of 9,658 (1.2%) Ashkenazi Jewish individuals from the general population (Couch et al. 1996; Roa et al. 1996; Neuhausen et al. 1996; Oddoux et al. 1996; Struewing et al. 1997; Satagopan et al. 2002; King et al. 2003). Additionally, the p.Ser1982ArgfsTer22 variant has been identified in a compound heterozygous state in four individuals, including two cousins, with Fanconi anemia and brain tumors from three different Ashkenazi Jewish families (Offit et al. 2003; Alter et al. 2007). The p.Ser1982ArgfsTer22 variant was absent from at least 1,726 non-Ashkenazi Jewish controls and is reported at a frequency of 0.00048 in the European (non-Finnish) population of the Exome Aggregation Consortium. Functional studies demonstrated decreased cell viability and survival in carriers of the p.Ser1982ArgfsTer22 variant (Wu et al. 2005). The variant protein was shown to be localized in the cytoplasm and not in the nucleus. The p.Ser1982ArgfsTer22 variant is predicted to result in truncation of approximately 41% of the BRCA2 protein, which would remove two signals required for nuclear localization and represents a likely mode of pathogenicity for the variant (Spain et al. 1999). In a study in mouse embryonic stem cells, the p.Ser1982ArgfsTer22 variant failed to rescue the loss of endogenous BRCA2 (Kuznetsov et al. 2008). Based on the collective evidence, the p.Ser1982ArgfsTer22 variant is classified as pathogenic for BRCA2-related disorders. This variant was observed by ICSL as part of a predisposition screen in an ostensibly healthy population.
Variant summary: BRCA2 c.5946delT (p.Ser1982ArgfsX22) results in a premature termination codon, predicted to cause a truncation of the encoded protein or absence of the protein due to nonsense mediated decay, which are commonly known mechanisms for disease. The variant allele was found at a frequency of 0.0002765 in 282088 control chromosomes (gnomAD), which does not exceed the estimated maximal expected allele frequency of a pathogenic BRCA2 variant (0.0007503) and was predominantly observed in the Ashkenazi Jewish subpopulation. This variant is a well-established Ashkenazi Jewish founder mutation. The variant, c.5946delT, has been reported in the literature in multiple individuals affected with Hereditary Breast and Ovarian Cancer (Abeliovich_1997, Friedman_1998). These data indicate that the variant is very likely to be associated with disease. At least one publication reports experimental evidence evaluating an impact on protein function (Wu_2005). Multiple ClinVar submissions after 2014 cite the variant as pathogenic. Based on the evidence outlined above, the variant was classified as pathogenic.
"This variant has been reported in ClinVar as Pathogenic (67 clinical laboratories) and as pathogenic (1 clinical laboratories) and as Pathogenic by ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel, ClinGen expert panel."
COSMIC Somatic Evidence
Open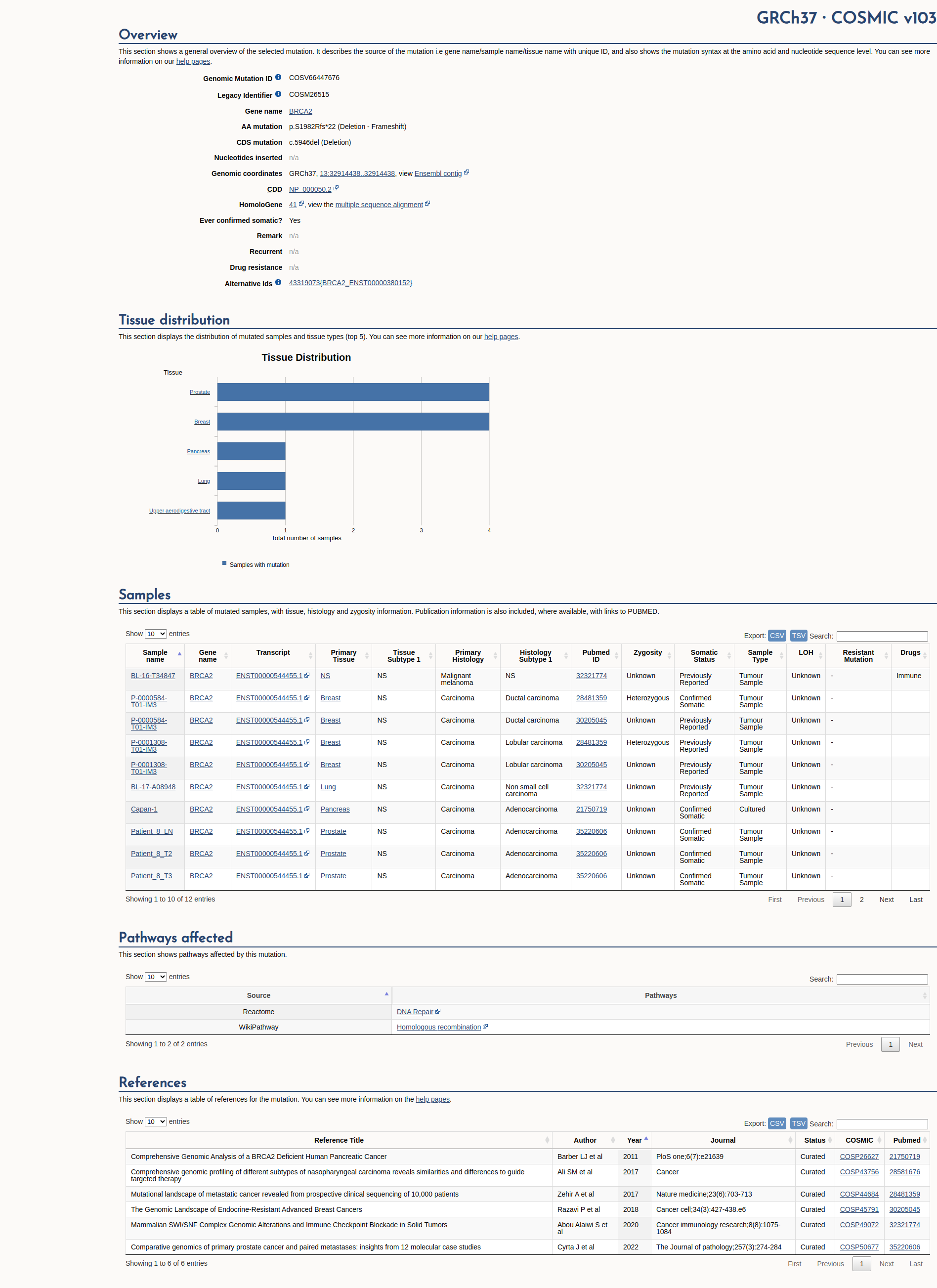
Functional Impact & Domains
Functional Domain
Error in OpenAI Consolidation. OncoKB: BRCA2S1982Rfs*22BRCA2S1982Rfs*22SomaticNCBI Gene:675|Show additional gene information Variant OverviewBRCA2, a tumor suppressor involved in the DNA damage response, is mutated in various cancer types.The BRCA2 S1982Rfs*22 is a truncating mutation in a tumor suppressor gene, and therefore is likely oncogenic.Hide mutation effect description The mutation effect description for truncating mutations in BRCA2 is: Truncating mutations of BRCA2 can produce several forms of C-terminally truncated BRCA2 proteins. Protein domains that are deleted in BRCA2 truncating mutations include the C-terminal DNA binding domain, the nuclear localization signal and the CDK2 phosphorylation site, the latter which binds RAD51 (PMID: 20878484, 24312913). Experimental studies have shown that truncating mutations impair nuclear localization of BRCA2, essential for normal BRCA2 function (PMID: 10570174). BRCA2 is essential for maintaining the integrity of homologous recombination during DNA damage response (PMID: 11239455). Inactivating or truncating mutations of this gene are associated with a significant increase in lifetime risk for the development of breast, ovarian, prostate or pancreatic cancers (PMID: 22193408). JAX-CKB: No results found
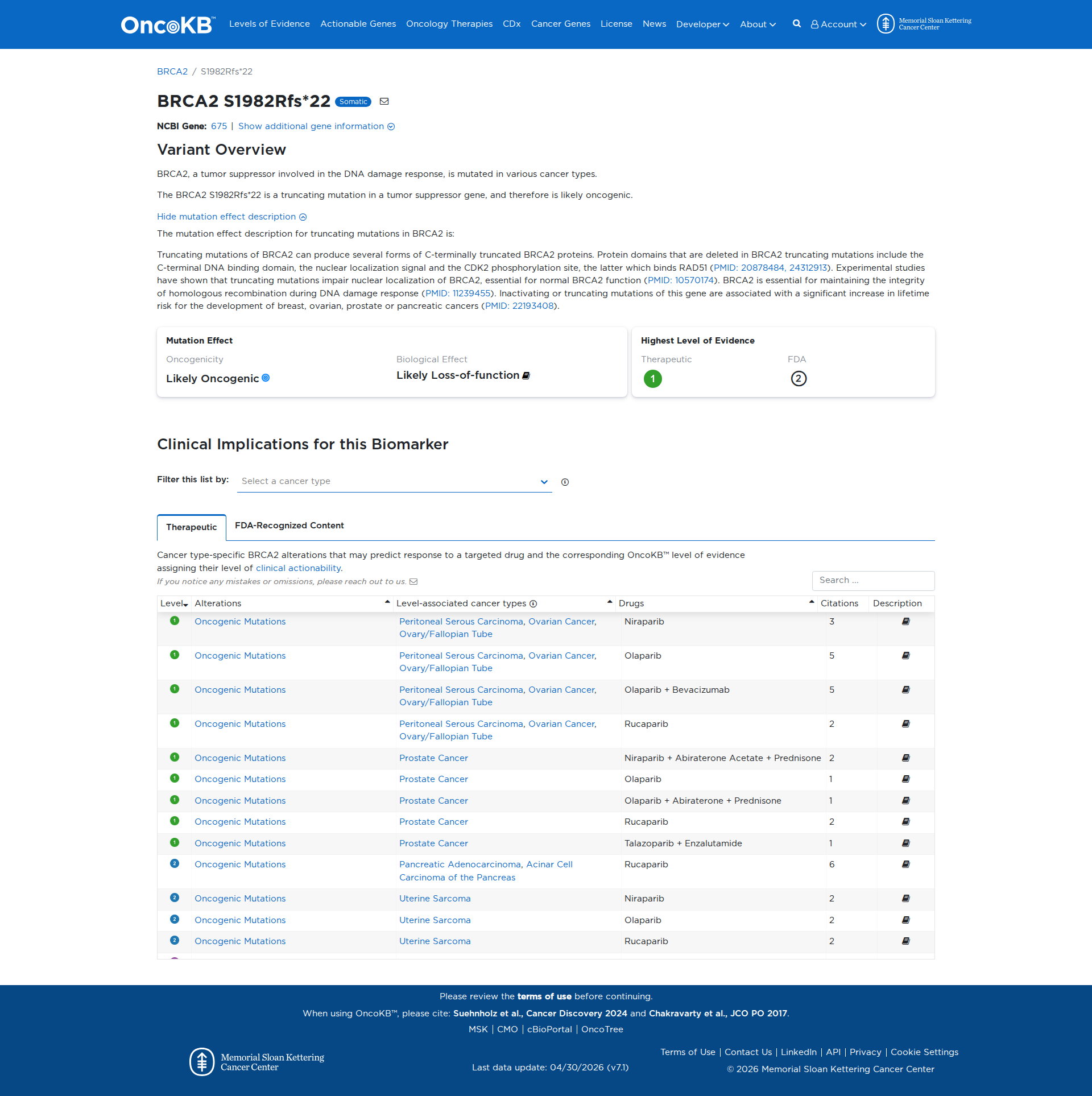
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.0 | -3 bp |
| Donor Loss (DL) | 0.0 | 202 bp |
| Acceptor Gain (AG) | 0.0 | -8 bp |
| Donor Gain (DG) | 0.0 | -445 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PVS1 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PS3 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PM2 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PP5 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)