Genetic Information
Gene & Transcript Details
| ID | Status | Details |
|---|---|---|
| NM_001330437.2 | Alternative | 6085 nt | 166–1959 |
| NM_001330437.1 | Alternative | 6151 nt | 215–2008 |
Variant Details
Clinical & Population Data
Population Frequency
gnomADClinVar
OpenVariant summary: The c.188A>G variant affects a conserved nucleotide, resulting in amino acid change from Tyr to Cys. 4/4 in-silico tools predict damaging outcome for this variant (SNPs&GO not captured due to low reliability index). This variant is found in 1/121890 control chromosomes at a frequency of 0.0000082, which does not exceed maximal expected frequency of a pathogenic allele (0.0000625). The variant has been reported in numerous affected individuals and families in the literature and has been shown to segregate with disease in affected families. The variant is considered a common pathogenic variant. In addition, multiple reputable clinical laboratory and database classified this variant as Pathogenic. Taken together, this variant was classified as Pathogenic.
This variant is classified as Pathogenic. Evidence in support of pathogenic classification: Variant is present in gnomAD (v2) <0.001 for a dominant condition (3 heterozygotes, 0 homozygotes); The variant has strong previous evidence of pathogenicity in unrelated individuals with Noonan syndrome. It is a 3-star pathogenic variant in ClinVar (reviewed by the ClinGen RASopathy Variant Curation Expert Panel), and is reported in the literature in individuals with Noonan syndrome (PMIDs: 11704759; 12325025; 12634870); The variant has strong evidence for segregation with disease in multiple families (PMID: 12325025; 12634870); Variant is located in a hotspot region or cluster of pathogenic variants (N-SH2 domain; DECIPHER); Missense variant consistently predicted to be damaging by multiple in silico tools and is highly conserved with a major amino acid change. Additional information: Variant is predicted to result in a missense amino acid change from tyrosine to cysteine; This variant is heterozygous; This gene is known to be associated with autosomal dominant disease; Both loss of function and gain of function are known mechanisms of disease for this gene. Metachondromatosis (MIM#156250), and Noonan syndrome with multiple lentigines have been associated with loss of function variants, whereas Noonan syndrome 1 (MIM#163950) is caused by gain of function variants (PMIDs: 11992261, 24935154, 21533187); Variants in this gene are known to have variable expressivity (PMID: 20301303); This variant has been shown to be maternally inherited by trio analysis.
This sequence change replaces tyrosine, which is neutral and polar, with cysteine, which is neutral and slightly polar, at codon 63 of the PTPN11 protein (p.Tyr63Cys). This variant is present in population databases (rs121918459, gnomAD 0.006%). This missense change has been observed in individuals with Noonan syndrome (PMID: 11704759, 11992261, 12325025, 12960218, 16498234, 21407260). It has also been observed to segregate with disease in related individuals. ClinVar contains an entry for this variant (Variation ID: 13333). Invitae Evidence Modeling of protein sequence and biophysical properties (such as structural, functional, and spatial information, amino acid conservation, physicochemical variation, residue mobility, and thermodynamic stability) indicates that this missense variant is expected to disrupt PTPN11 protein function with a positive predictive value of 95%. Experimental studies have shown that this missense change affects PTPN11 function (PMID: 22711529). For these reasons, this variant has been classified as Pathogenic.
Variant classified using ACMG guidelines
The p.Tyr63Cys variant in PTPN11 has been reported in >40 individuals with Noona n syndrome, occurred de novo in some sporadic cases and segregated with disease in numerous families (Tartaglia 2002, Kosaki 2002, Maheshwari 2002, Musante 2003 , Loh 2004, Kratz 2005, Takahashi 2006, Becker 2007, Jongmans 2011, Simsek-Kiper 2012, LMM data). In addition, this variant has been identified as a somatic var iant in one individual with chronic myelomonocytic leukemia (CMML; Loh 2004) and as a germline variant in two individuals with both clinical features of Noonan syndrome and a malignancy (precursor B-ALL and basal cell carcinoma; Jongmans 20 11). This variant has also been identified in 1/17248 East Asian chromosomes and 1/33576 Latino chromosomes by gnomAD (http://gnomad.broadinstitute.org). Moreov er, the p.Tyr63Cys variant has been classified as pathogenic on April 3, 2018 by the ClinGen-approved RASopathy Expert Panel (ClinVar SCV000616372.1). In summar y, this variant meets criteria to be classified as pathogenic for Noonan syndrom e in an autosomal dominant manner based upon presence in multiple affected indiv iduals, de novo occurrences and segregation studies. ACMG/AMP Criteria applied: PS4, PP1_Strong, PM6_Strong.
"This variant has been reported in ClinVar as Pathogenic (50 clinical laboratories) and as pathogenic (1 clinical laboratories) and as Uncertain significance (1 clinical laboratories) and as Pathogenic by ClinGen RASopathy Variant Curation Expert Panel expert panel."
COSMIC Somatic Evidence
Open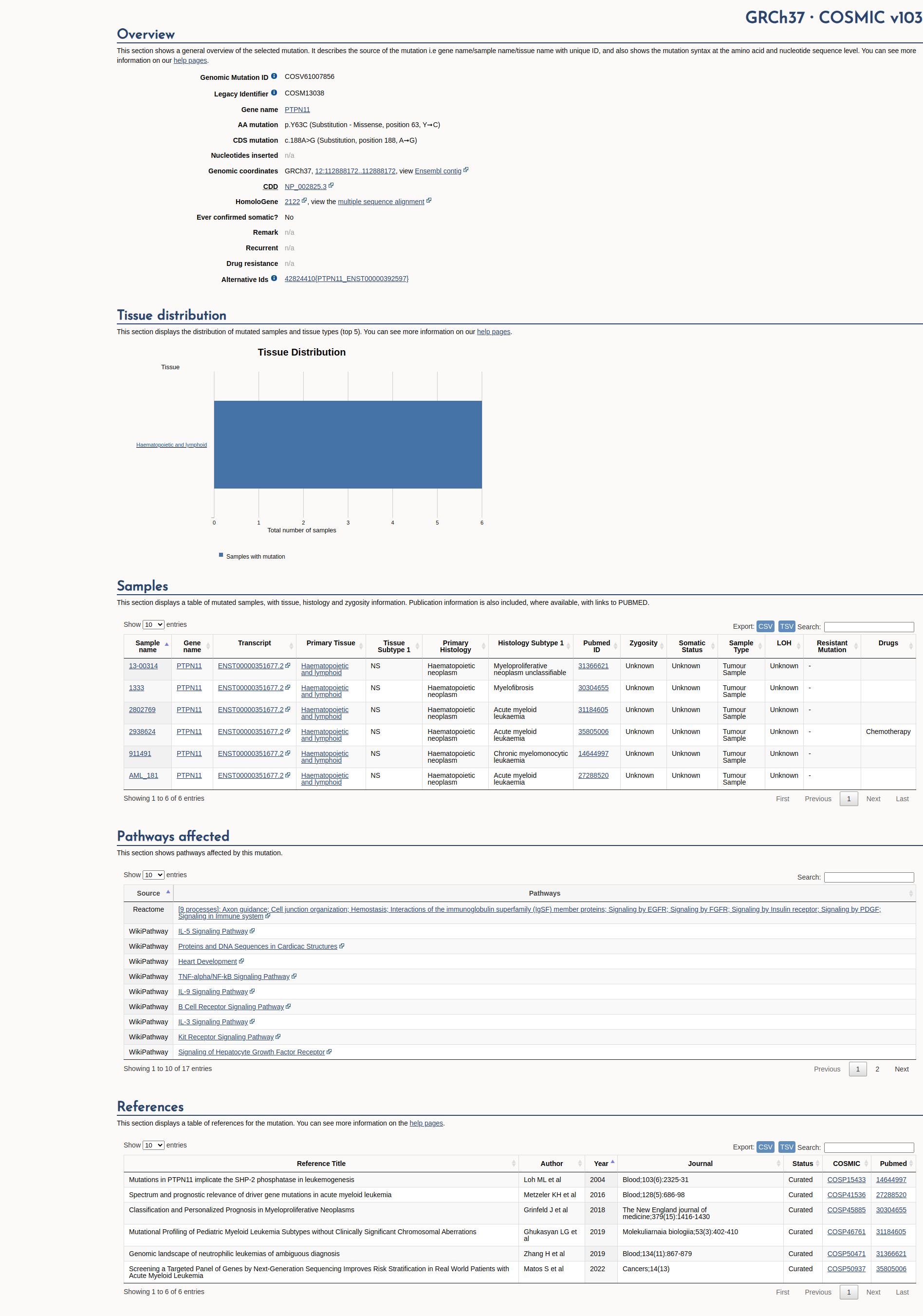
Functional Impact & Domains
Functional Domain
Error in OpenAI Consolidation. OncoKB: PTPN11Y63CPTPN11Y63CSomaticNCBI Gene:5781|Show additional gene information Variant OverviewPTPN11, a protein tyrosine phosphatase, is altered in various solid and hematologic malignancies.The PTPN11 Y63C mutation is likely oncogenic.Hide mutation effect description The PTPN11 Y63C mutation is located in its N-SH2 domain. Cells transfected with Y63C mutant alleles show a modest increase in phosphatase activity (PMID: 15834506). JAX-CKB: No results found
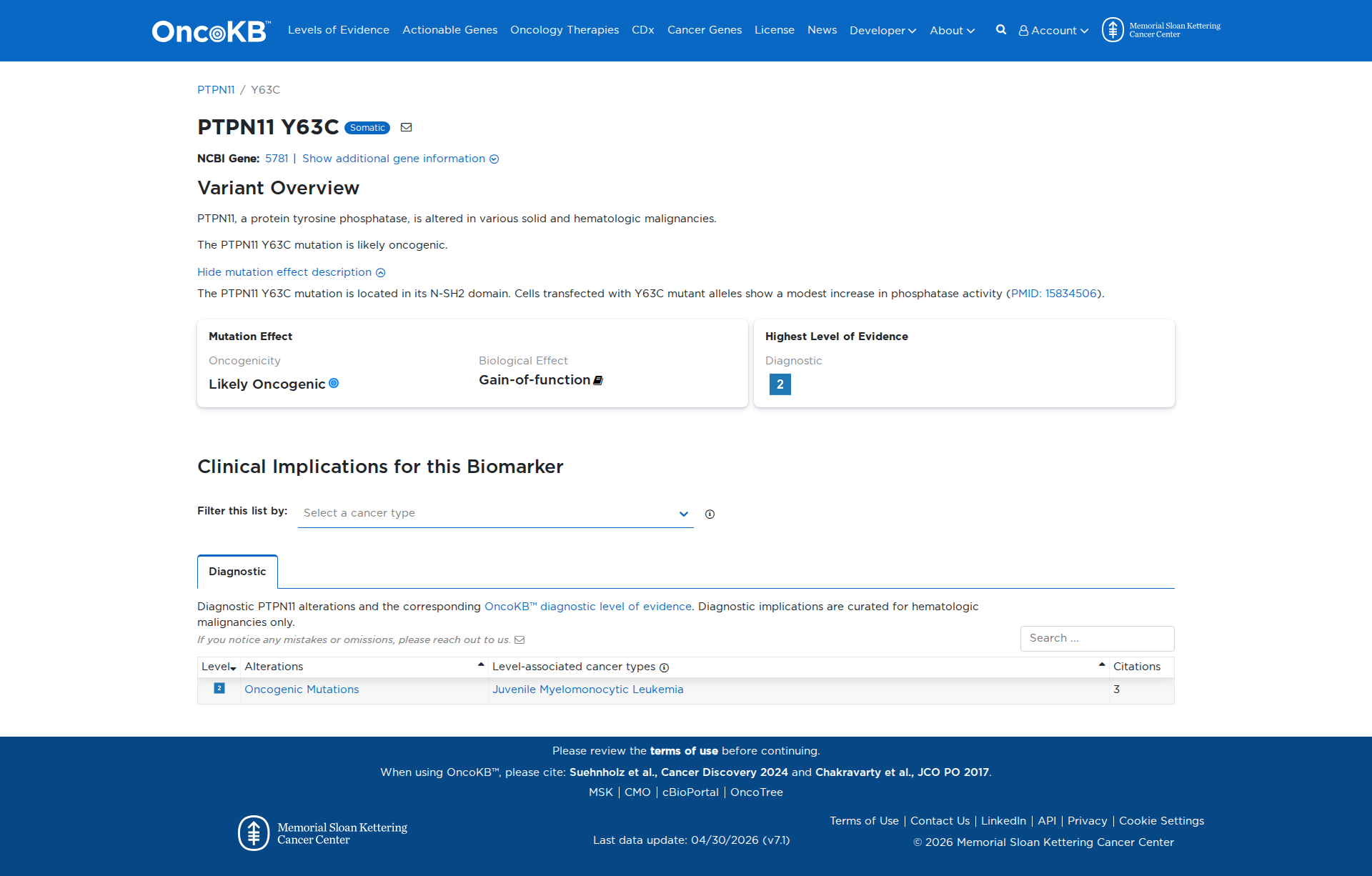
Click on previews to view full database entries. External databases may require institutional access.
Computational Analysis
Pathogenicity Predictions
SpliceAISpliceAI Scores
Window: ±500bp| Effect Type | Score | Position |
|---|---|---|
| Acceptor Loss (AL) | 0.03 | -139 bp |
| Donor Loss (DL) | 0.0 | -10 bp |
| Acceptor Gain (AG) | 0.03 | -47 bp |
| Donor Gain (DG) | 0.0 | -256 bp |
VCEP Guidelines
Applied ACMG/AMP Criteria (VCEP Specific)
PS3 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PM2 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
PP5 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)
BP4 (Unknown (Pre-LLM))
From pre-LLM assessment (LLM Failed)