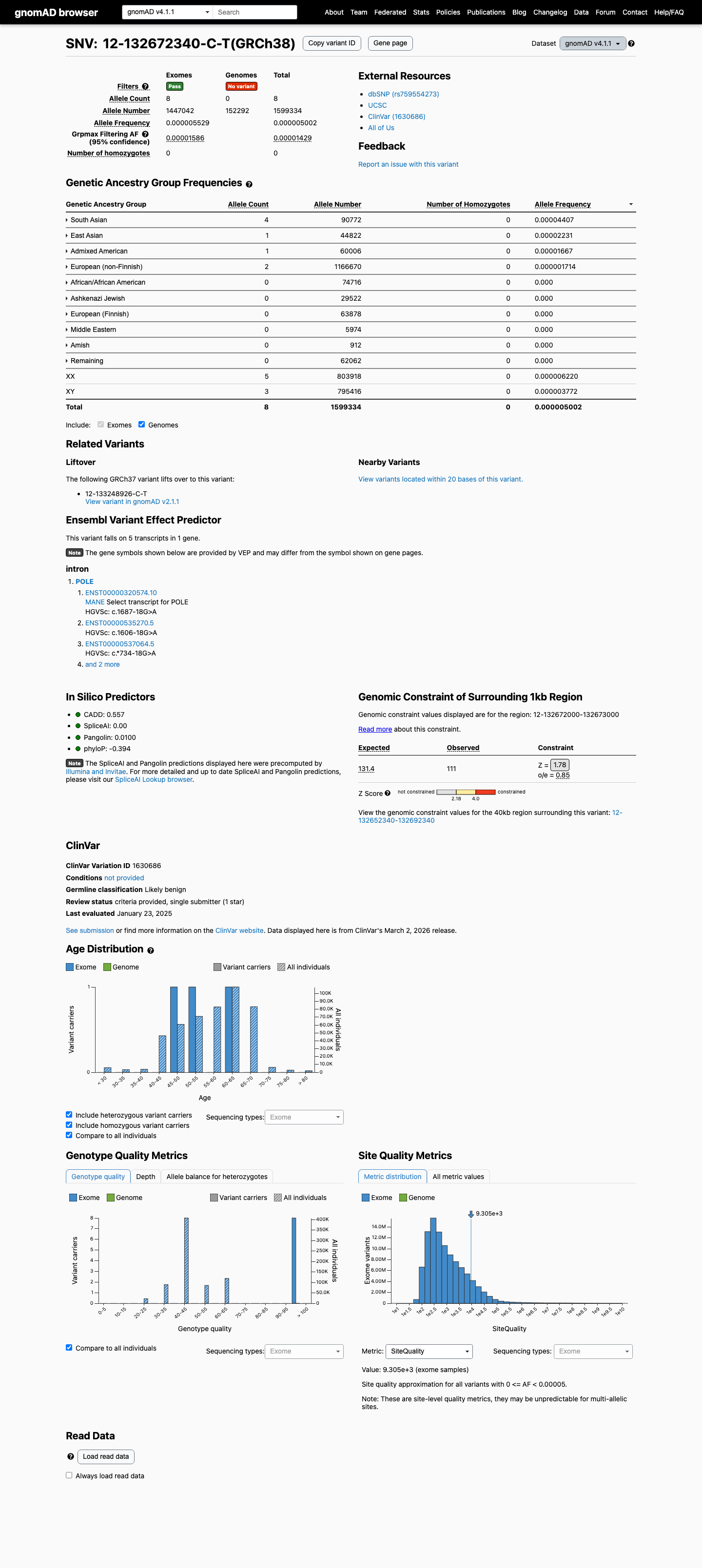
NM_006231.4:c.1687-18G>A is a noncanonical intronic POLE variant observed in gnomAD v4.1 at 8/1447042 alleles (AF 5.52852e-06; 0.00055%), with the highest frequency in South Asians at 4/85948 alleles (AF 4.65398e-05; 0.00465%), which is below the non-VCEP PM2 threshold of 0.1%.
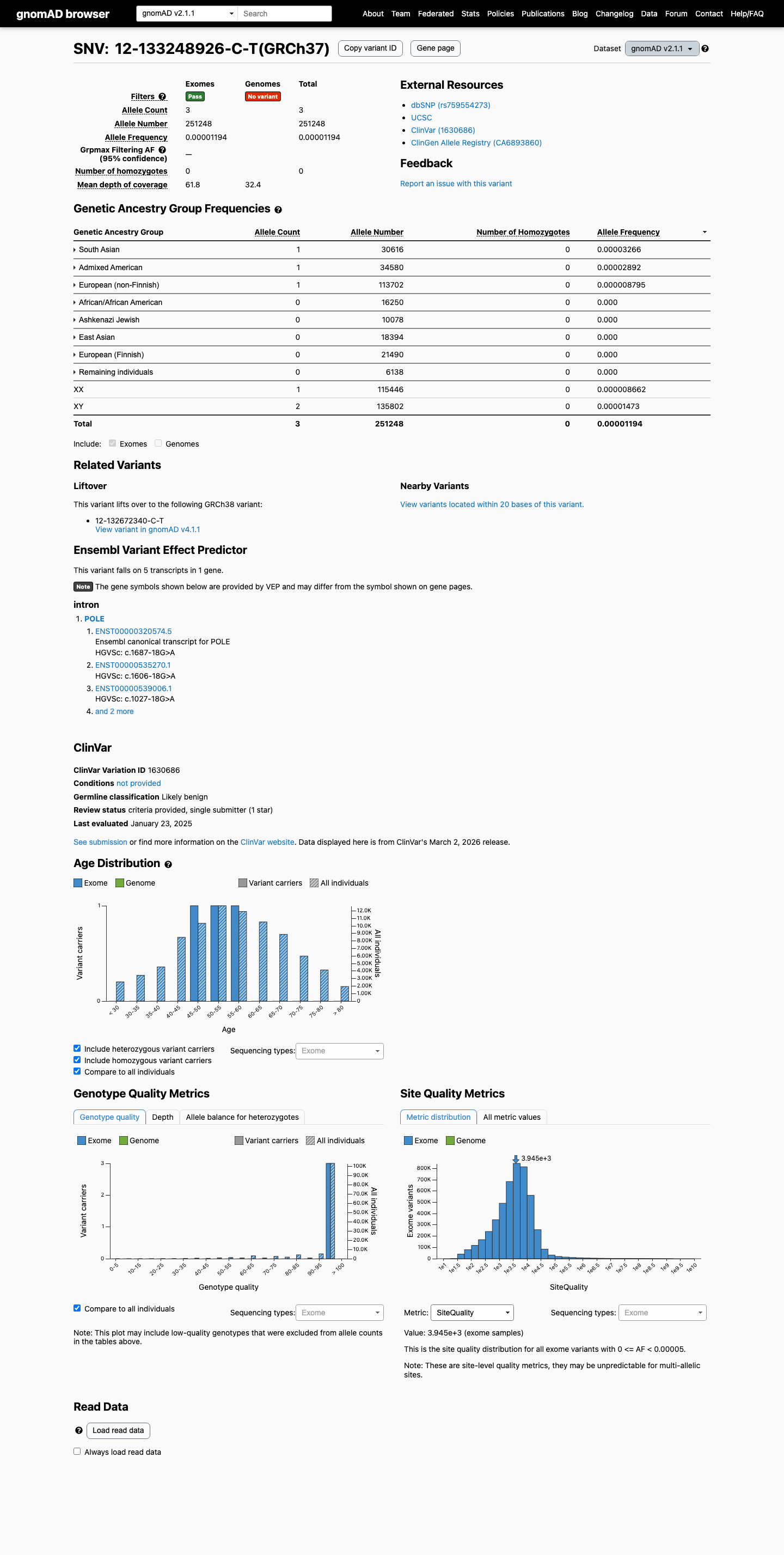
gnomad_v4 ↗Population data are similarly rare in gnomAD v2.1 at 3/251248 alleles (AF 1.19404e-05; 0.00119%), with the highest South Asian frequency at 1/30616 alleles (AF 3.26627e-05; 0.00327%), and these values also remain below the BS1 threshold of 0.3% and BA1 threshold of 1%.
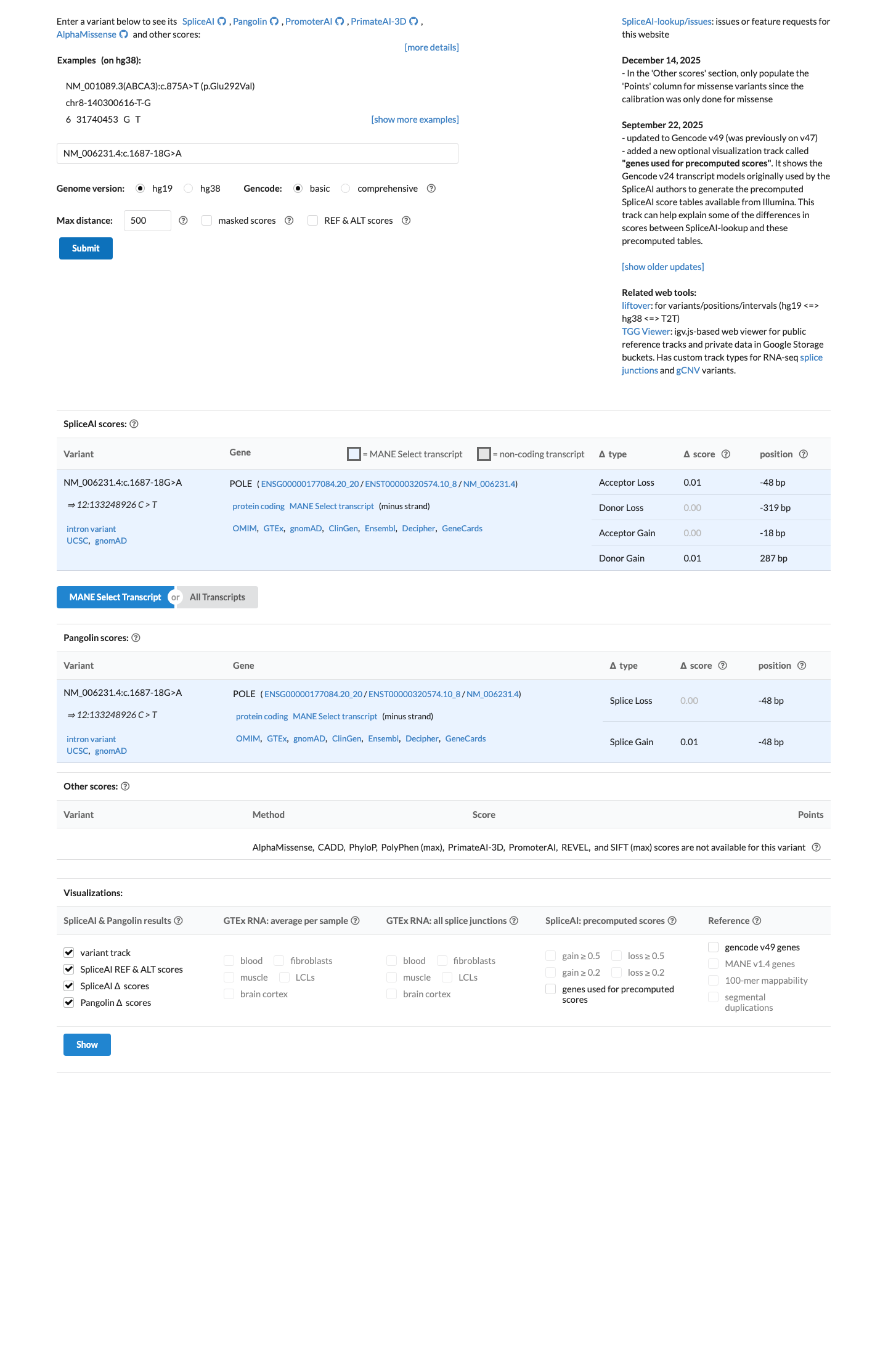
gnomad_v2 ↗SpliceAI predicts a maximum delta score of 0.01, which is below the 0.2 threshold commonly used to indicate splice-impact concern and supports a benign computational assessment under BP4 rather than pathogenic computational evidence.
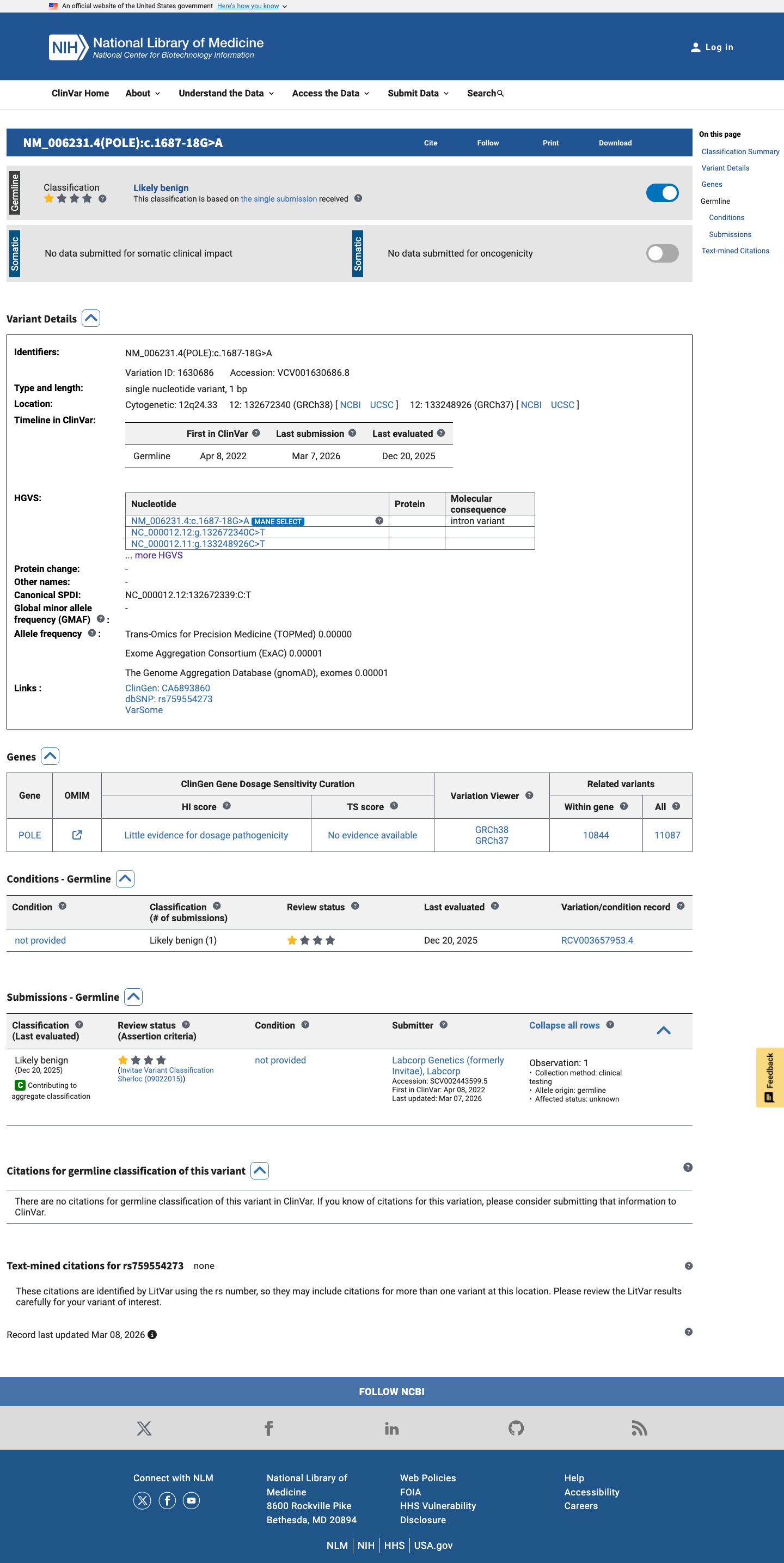
spliceai ↗ClinVar contains a single submitter classification of Likely benign, which is directionally consistent with the splice prediction but is not used here as a standalone ACMG criterion.
clinvar ↗With conflicting supporting evidence from rarity and benign computational data, and without stronger pathogenic or benign evidence, NM_006231.4:c.1687-18G>A is classified as a Variant of Uncertain Significance.