NM_000051.4:c.8261C>T (p.Thr2754Ile) is a missense variant in ATM, a gene for which loss of function is an established mechanism of disease in ataxia-telangiectasia and in which germline variants confer susceptibility to breast, ovarian, and pancreatic cancer (ClinGen HBOP VCEP v1.5.0).1 This variant is present at extremely low frequency in population databases: gnomAD v4.1 reports 4 heterozygous carriers among 1,605,254 alleles (total AF=2.49e-06; grpmax FAF=2.8e-07) with no homozygotes observed, and gnomAD v2.1 reports 1 heterozygous carrier among 31,352 alleles (AF=3.19e-05). It is absent from gnomAD-Canada v1.0.2 In silico predictors are inconclusive: REVEL score is 0.633 (below the ATM VCEP PP3 threshold of >0.7333 but above the BP4 threshold of ≤0.249), BayesDel score is -0.0763065, and SpliceAI predicts no significant splice impact (max delta score = 0.01).3 This variant has been reported in ClinVar as Uncertain Significance by 6 clinical laboratories (ClinVar Variation ID: 127457), with no expert panel classifications available. No functional studies, case-control analyses, segregation data, or observations in trans with pathogenic ATM variants have been identified for this specific variant.4 The variant is absent from COSMIC (no somatic cancer reports) and does not fall within a statistically significant Cancer Hotspot. OncoKB reports no reviewed functional evidence and classifies this variant as having Unknown Oncogenic Effect.5 Under the ClinGen HBOP VCEP for ATM v1.5.0, only PM2_Supporting is met (allele frequency ≤0.001% in gnomAD v4). All other applicable criteria are either not met or not applicable. With a single supporting-level pathogenic criterion and no benign criteria met, the variant is classified as Uncertain Significance (VUS) per the ACMG/AMP 2015 combining rules as adopted by the ATM VCEP.6
ATM
Final classification
VUS
ATM c.8261C>T · p.Thr2754Ile
ATM
NM_000051.4:c.8261C>T (p.Thr2754Ile) is a missense variant in ATM, a gene for which loss of function is an established mechanism of disease in ataxia-telangiectasia and in which germline variants confer susceptibility to breast, ovarian, and pancreatic cancer (ClinGen HBOP VCEP v1.5.0).
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework was evaluated deterministically with applied criteria: PM2 supporting; no rule matched the adjudicated criteria.
Classification rationale
PM2
VUS
ATM c.8261C>T
PM2
→
VUS
1
cspec ↗pvs1_gene_context
3
revelbayesdelspliceai ↗
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 11 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in gnomAD v4.1 (total AF=2.49e-06; 4/1,605,254 alleles; grpmax FAF=2.8e-07; 0 homozygotes), meeting the ATM VCEP PM2_Supporting threshold of ≤0.001% (≤0.00001) in the gnomAD v4 dataset.
gnomAD v4.1: total AF=2.49e-06 (0.000249%)4 alleles in 1605
Assessed · not applied
Pathogenic
PS1
PS1 requires a known pathogenic missense variant at the same amino acid residue (p.Thr2754) with a different nucleotide change, and splicing must be ruled out for both.
PS3
No variant-specific functional assay data are available for NM_000051.4:c.8261C>T (p.Thr2754Ile).
PS4
The ATM VCEP requires a formal case-control study with p-value ≤0.05 and an odds ratio/hazard ratio/relative risk ≥2 or lower 95% CI ≥1.5.
PM3
No evidence of this variant detected in trans with a known pathogenic ATM variant in an individual with ataxia-telangiectasia (A-T) was identified.
PP1
No cosegregation data are available for this variant.
PP3
The REVEL score for this missense variant is 0.633, which is below the ATM VCEP PP3 threshold of >0.7333.
Benign
BA1
The ATM VCEP BA1 threshold requires a grpmax filtering allele frequency >0.5% in gnomAD v4.
BS1
The ATM VCEP BS1 threshold requires a grpmax filtering allele frequency >0.05% in gnomAD v4.
BS3
No functional evidence demonstrating that this variant rescues ATM-specific function (e.g., phosphorylation of ATM-specific targets) or radiosensitivity has been identified.
BP2
No evidence of this variant occurring in trans with a known pathogenic ATM variant in an unaffected (non-A-T) individual has been identified.
BP4
The ATM VCEP BP4 missense threshold requires a REVEL score ≤0.249.
N/A · 16
PVS1 · PS2 · PM1 · PM4 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency
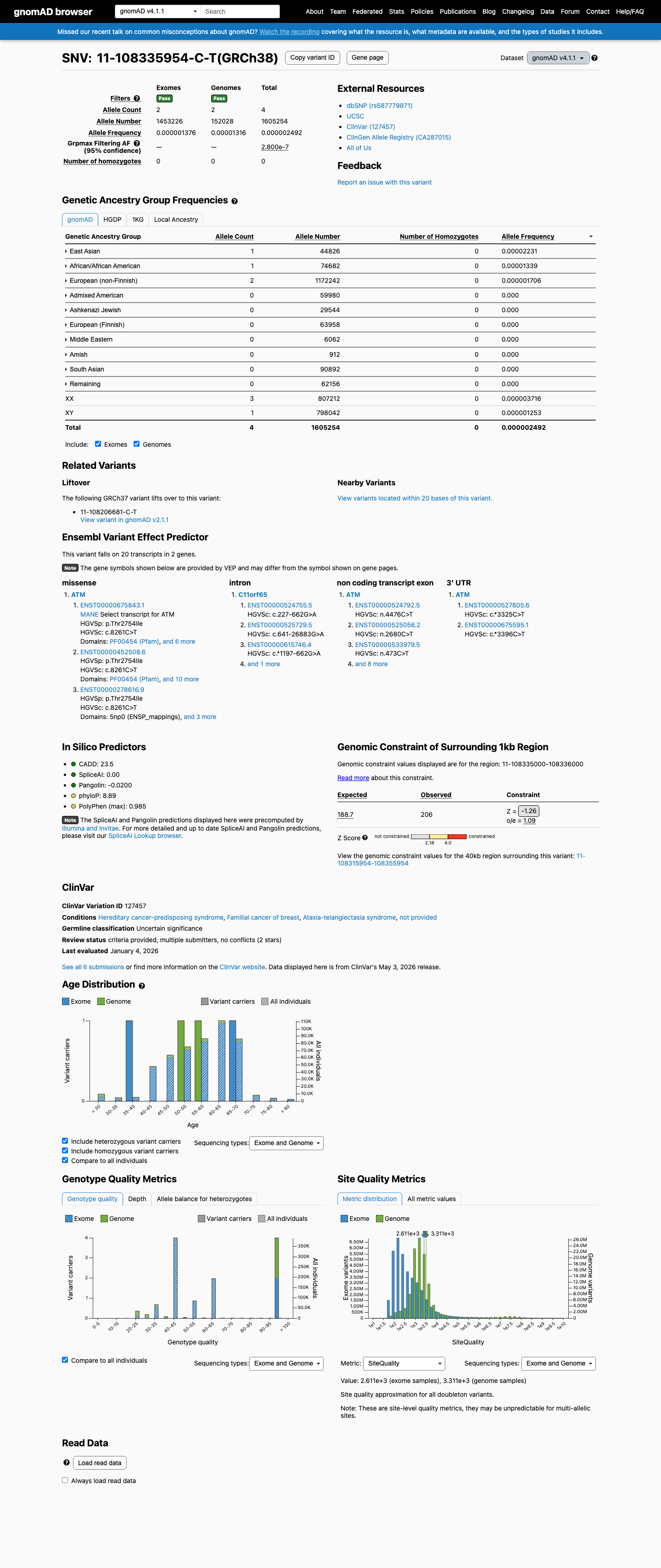
gnomAD v4.1
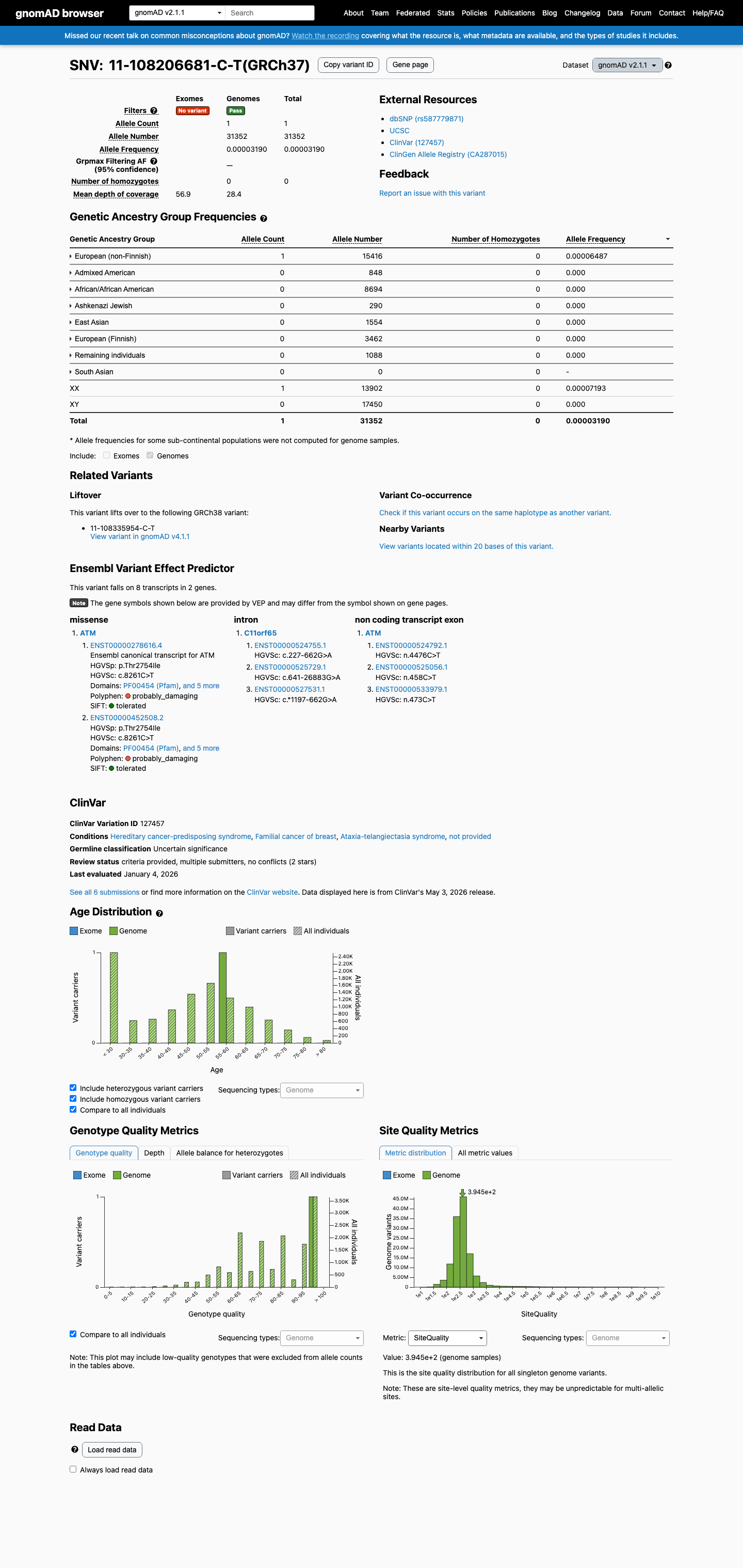
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.49182e-06; MAF= 0.00025%, 4/1605254 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 2.23085e-05; MAF= 0.00223%, 1/44826 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.18959e-05; MAF= 0.00319%, 1/31352 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 6.48677e-05; MAF= 0.00649%, 1/15416 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,605,254
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
East Asian 1 / 44,826 |
0.0022% |
African/African American 1 / 74,682 |
0.0013% |
European (non-Finnish) 2 / 1,172,242 |
0.00017% |
+ 7 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0032%
· 1 / 31,352
0 hom
0 hom
European (non-Finnish) 1 / 15,416 |
0.0065% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
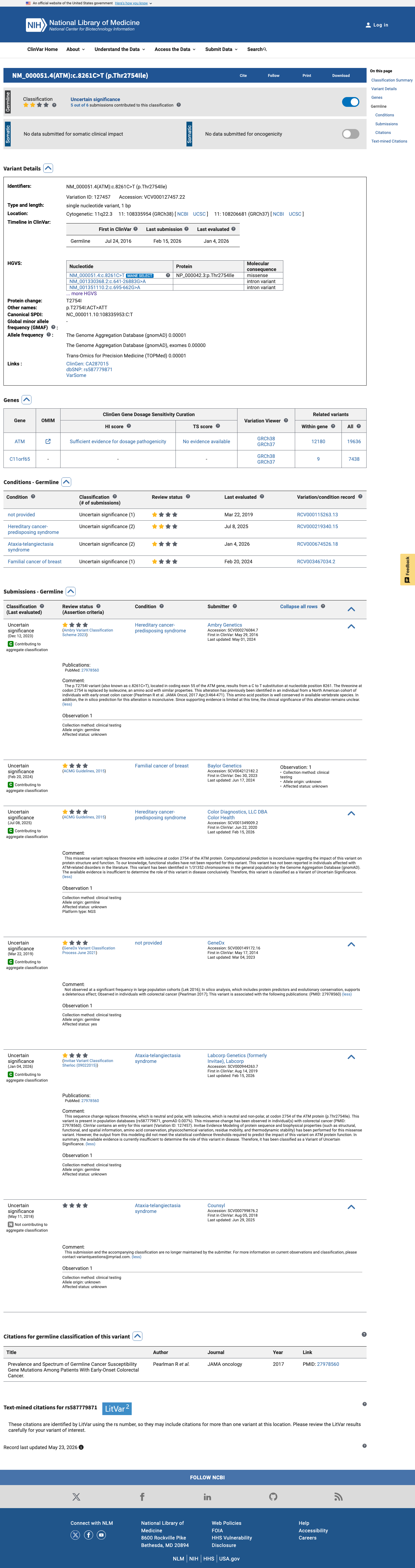
ClinVar
This variant has been reported in ClinVar as Uncertain significance (6 clinical laboratories). (ClinVarID = 127457)
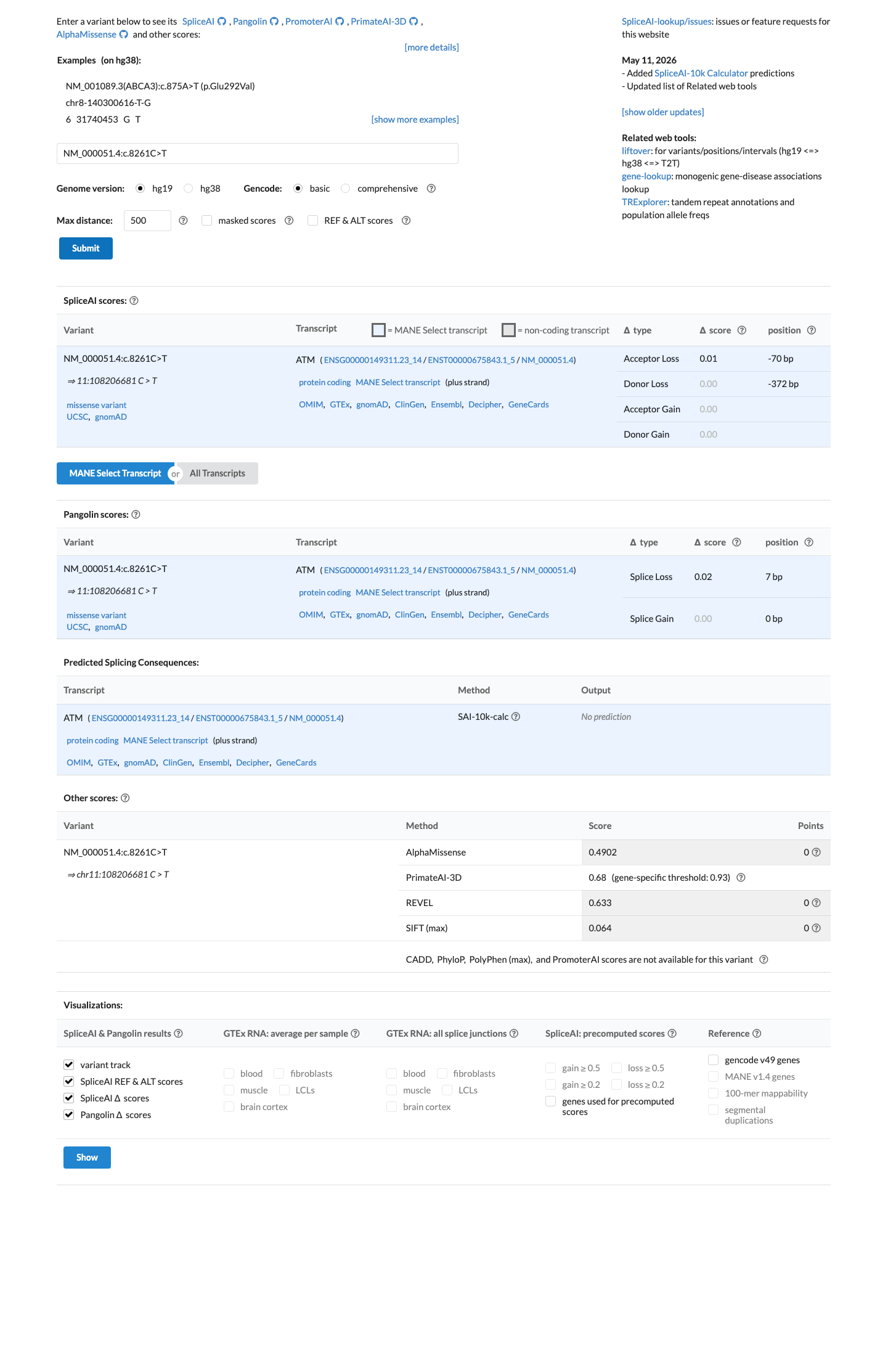
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.633. BayesDel score = -0.0763065.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. ATM, a kinase involved in the DNA damage response, is mutated in various solid and hematologic malignancies.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 10 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
27978560 ↗
Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer.
CLINVAR
34242744 ↗
Customizing local and systemic therapies for women with early breast cancer: the St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
17508274 ↗
Risk assessment and genetic counseling for hereditary breast and ovarian cancer: recommendations of the National Society of Genetic Counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR