NM_004168.4:c.163T>C (p.Tyr55His) is classified as Benign based on the ACMG/AMP 2015 framework.1 This variant meets BA1 (stand-alone benign): the allele frequency in the East Asian population is 1.96% in gnomAD v2.1 (391/19,954 alleles, 5 homozygotes) and 2.39% in gnomAD v4.1 (1,074/44,890 alleles, 10 homozygotes), with a gnomAD v4.1 grpmax FAF of 2.27%. An allele frequency exceeding 1% in a general population is inconsistent with a role in rare Mendelian disease.2 Additional benign evidence includes BS1 (East Asian AF well above 0.3%), BS2 (12 homozygotes observed in gnomAD v4.1 in a gene where biallelic loss-of-function causes severe early-onset recessive disease), BP4 (REVEL = 0.392; BayesDel = 0.00237; SpliceAI max delta = 0.03; all predictors converge on a benign interpretation), and BP6 (ClinVar consensus of Benign/Likely benign from 11 clinical laboratories).3 No pathogenic criteria are met. PVS1 is not applicable (missense variant). PP3 is not met (in silico predictors favor benign). PM1 is not met (the variant is common in population databases despite lying in the FAD-binding domain). PS4 is not met (the variant is classified as benign across clinical laboratories and observed at appreciable frequency in unaffected populations).4 The classification of Benign is driven by BA1, which alone is sufficient to classify a variant as Benign under the ACMG/AMP 2015 combination rules.5
SDHA
Final classification
Benign
SDHA c.163T>C · p.Tyr55His
SDHA
NM_004168.4:c.163T>C (p.Tyr55His) is classified as Benign based on the ACMG/AMP 2015 framework.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 supporting benign, BP4 supporting benign, BP6 supporting benign; combination = 1 stand-alone benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP4BP6
Benign
SDHA c.163T>C
BA1 + BS1 + BS2 + BP4 + BP6
→
Benign
3
gnomad_v2 ↗gnomad_v4 ↗revelbayesdelspliceai ↗clinvar ↗
4
spliceai ↗revelbayesdelgnomad_v2 ↗gnomad_v4 ↗clinvar ↗
Gene diagram
· NM_004168.4 · variants mapped to exon structure
SDHA
NM_004168.4
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 19 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
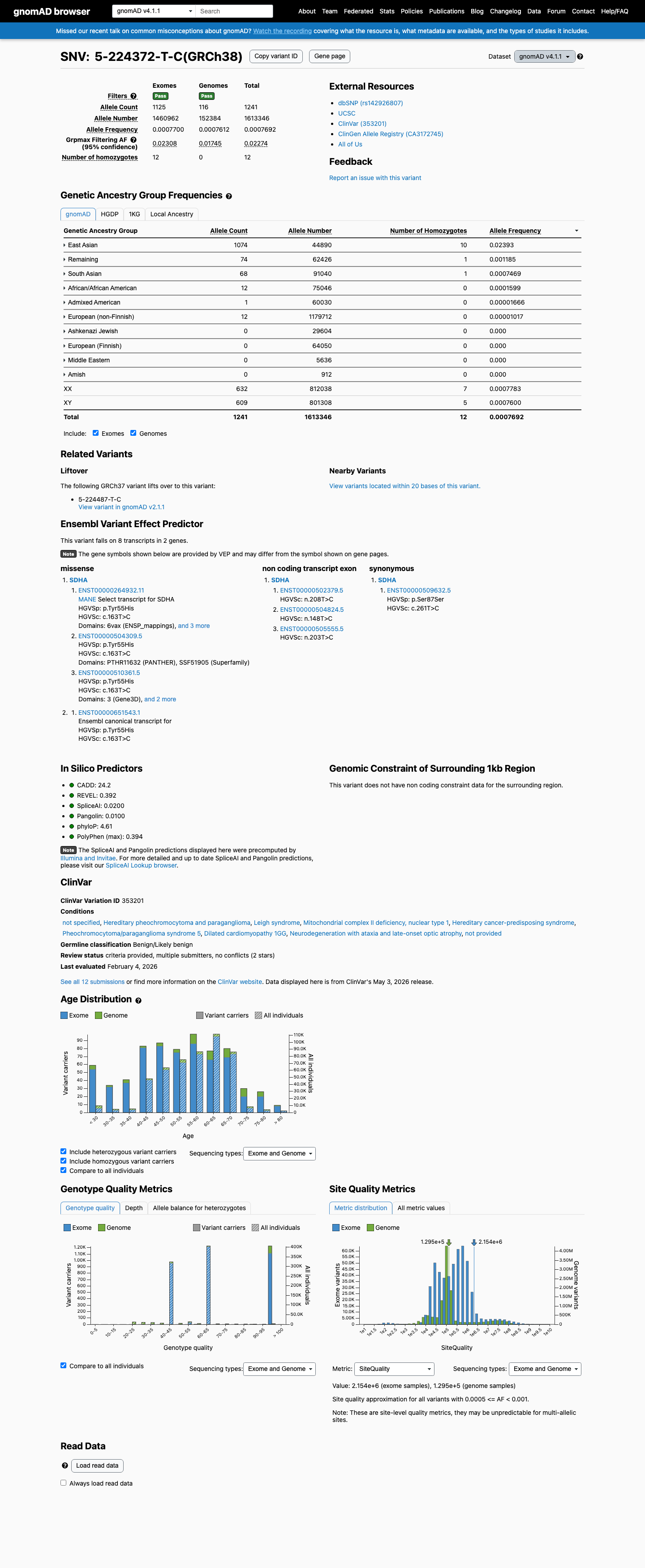
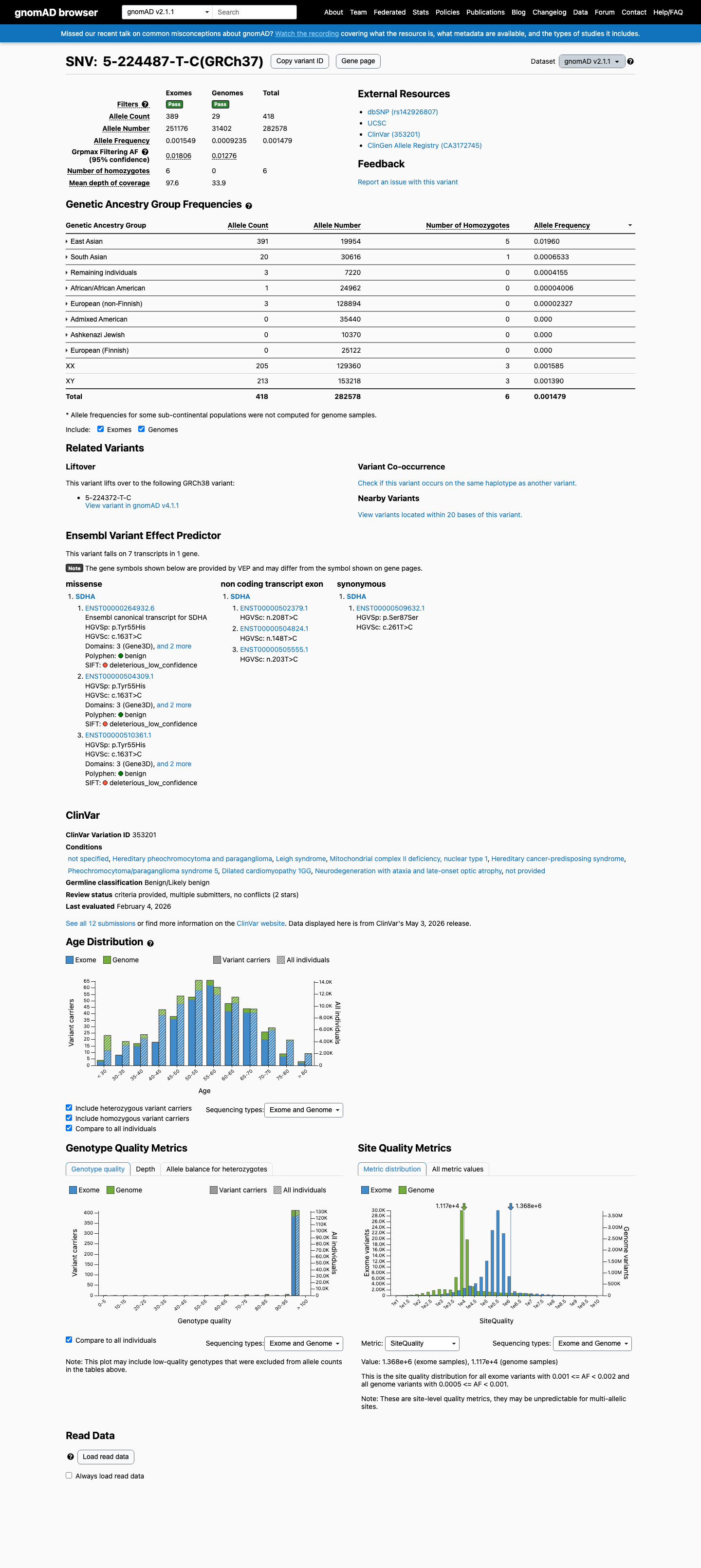
This variant has an allele frequency exceeding 1% in the East Asian population across multiple population databases: gnomAD v2.1 East Asian AF = 1.96% (391/19,954 alleles, 5 homozygotes), gnomAD v4.1 East Asian AF = 2.39% (1,074/44,890 alleles, 10 homozygotes), and gnomAD-Canada v1.0 East Asian AF = 1.12% (15/1,338 alleles). The gnomAD v4.1 grpmax FAF is 2.27%, well above the 1% BA1 threshold. This allele frequency is too high to support a causative role in rare Mendelian disease, meeting BA1 (stand-alone benign).
gnomAD v2.1 EAS AF = 1.96%5 homozygotesgnomAD v4.1 EAS AF = 2.39%
✓
BS1
strong
Benign
The allele frequency in the East Asian population exceeds 0.3% in all queried population databases: gnomAD v2.1 EAS AF = 1.96%, gnomAD v4.1 EAS AF = 2.39%, gnomAD-Canada EAS AF = 1.12%. The overall frequency is also elevated in gnomAD v2.1 (0.148%) although below 0.3% in gnomAD v4.1 (0.077%). BS1 is met based on the East Asian subpopulation frequency, which is well above the 0.3% threshold. Note: BA1 (stand-alone benign) is also met; BS1 provides additional population-frequency support but is not counted independently when BA1 is applied.
gnomAD v2.1 EAS AF = 1.96% (>0.3%)gnomAD v4.1 EAS AF = 2.39% (>0.3%)gnomAD-Canada EAS AF = 1.12% (>0.3%)
✓
BS2
supporting
Benign
This variant has been observed in the homozygous state in multiple individuals across gnomAD databases: 6 homozygotes in v2.1 (exomes), 12 homozygotes in v4.1 (of which 10 are East Asian), and 0 homozygotes in gnomAD-Canada. SDHA biallelic loss-of-function causes severe, early-onset recessive conditions (mitochondrial complex II deficiency, Leigh syndrome) with expected full penetrance. The observation of 12 apparently healthy adult homozygotes in population databases is inconsistent with a highly penetrant recessive pathogenic variant. BS2 is met at the supporting benign level.
v2.1: 6 homozygotesv4.1: 12 homozygotes (10 in East Asian)Recessive SDHA disorders have full early penetrance
✓
BP4
supporting
Benign
Multiple in silico predictors consistently suggest a benign effect for this missense variant. REVEL score is 0.392 (below the commonly used 0.5 pathogenic threshold). BayesDel score is 0.00237 (very low, strongly favoring benign). SpliceAI predicts no significant splice alteration (max delta score = 0.03). The convergent computational evidence supports a lack of functional impact, meeting BP4 at the supporting benign level.
REVEL = 0.392 (<0.5benign)BayesDel = 0.00237 (benign)
✓
BP6
supporting
Benign
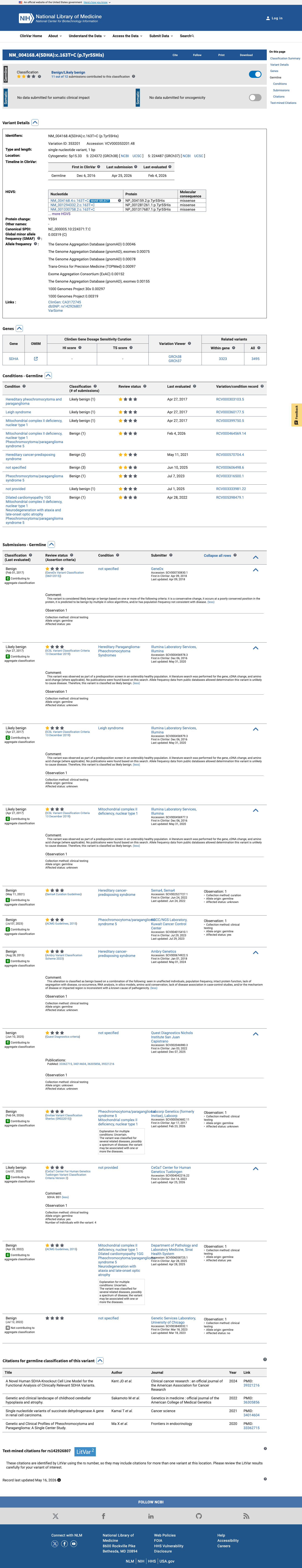
This variant has been reported in ClinVar as Benign by 6 clinical laboratories, Likely benign by 4 clinical laboratories, and benign by 1 clinical laboratory (ClinVar Variation ID: 353201). The aggregate ClinVar consensus is Benign/Likely benign with 11 submitting laboratories, though none represent an expert panel. BP6 is met at the supporting benign level based on the consistent benign classification across multiple clinical testing laboratories.
ClinVar: Benign (6 labs)Likely benign (4 labs)benign (1 lab)
Assessed · not applied
Pathogenic
PS1
No alternate nucleotide change at the same amino acid position with an established pathogenic classification was identified.
PS2
No de novo occurrence of this variant in a patient with disease and confirmed paternity/maternity was identified in the literature or databases.
PS3
No published functional studies specifically evaluating the functional impact of NM_004168.4:c.163T>C (p.Tyr55His) were identified.
PS4
No case-control study demonstrating a statistically significant enrichment of this variant in affected individuals compared to controls was identified.
PM1
Residue Tyr55 lies within the FAD-binding domain of SDHA (Rossmann-fold motif).
PM2
In gnomAD v4.1, the total allele frequency is 0.00077 (0.077%), which is below the 0.1% threshold.
PM3
No reports of this variant occurring in trans with a known pathogenic or likely pathogenic variant in a recessive SDHA-associated disorder (e.g., mitochondrial complex II deficiency, Leigh syndrome) were identified.
PM5
Automated PM5 candidate harvesting was unable to identify same-residue comparator variants with pathogenic classifications satisfying classic PM5 semantics.
PM6
No assumed de novo observation (without confirmation of paternity/maternity) was identified for this variant.
PP1
No family cosegregation data were identified for NM_004168.4:c.163T>C.
PP2
PP2 requires a low rate of benign missense variation and a high rate of pathogenic missense variants in the gene.
PP3
Multiple in silico predictors suggest a benign effect for this missense variant.
PP4
PP4 requires patient-specific phenotypic and family history information that is highly specific for the disease.
PP5
PP5 requires a reputable source to have recently reported the variant as pathogenic.
Benign
BS3
No well-established functional studies demonstrating a neutral (non-damaging) effect of NM_004168.4:c.163T>C (p.Tyr55His) were identified.
BS4
No evidence of non-segregation of this variant with disease in affected families was identified.
BP1
BP1 applies to missense variants in genes where primarily truncating variants are known to cause disease.
BP2
No observation of this variant in trans with a pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant in any inheritance pattern, was identified.
BP5
BP5 applies when an alternate molecular basis for disease has been identified in a case with a different pathogenic variant.
N/A · 4
PVS1 · PM4 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000769209; MAF= 0.07692%, 1241/1613346 alleles, homozygotes = 12) and has highest observed frequency in the East Asian population (AF= 0.0239252; MAF= 2.39252%, 1074/44890 alleles, homozygotes = 10); grpmax FAF= 0.0227368.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00147924; MAF= 0.14792%, 418/282578 alleles, homozygotes = 6) and has highest observed frequency in the East Asian population (AF= 0.0195951; MAF= 1.95951%, 391/19954 alleles, homozygotes = 5); grpmax FAF= 0.0180621.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00124891; MAF= 0.12489%, 23/18416 alleles, homozygotes = 0) and has highest observed frequency in the eas population (AF= 0.0112108; grpmax FAF95= 0.00690997).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.077%
· 1241 / 1,613,346
12 hom · FAF 2.3%
12 hom · FAF 2.3%
East Asian 1074 / 44,890 |
2.4% 10 hom |
Remaining individuals 74 / 62,426 |
0.12% 1 hom |
South Asian 68 / 91,040 |
0.075% 1 hom |
African/African American 12 / 75,046 |
0.016% |
Admixed American 1 / 60,030 |
0.0017% |
European (non-Finnish) 12 / 1,179,712 |
0.001% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.15%
· 418 / 282,578
6 hom · FAF 1.8%
6 hom · FAF 1.8%
East Asian 391 / 19,954 |
2% 5 hom |
South Asian 20 / 30,616 |
0.065% 1 hom |
Remaining individuals 3 / 7,220 |
0.042% |
African/African American 1 / 24,962 |
0.004% |
European (non-Finnish) 3 / 128,894 |
0.0023% |
+ 3 not observed (Admixed American, Ashkenazi Jewish, European (Finnish))
gnomAD Canada 🇨🇦
0.12%
· 23 / 18,416
0 hom · FAF 0.69%
0 hom · FAF 0.69%
East Asian 15 / 1,338 |
1.1% |
Remaining individuals 2 / 1,138 |
0.18% |
South Asian 2 / 1,362 |
0.15% |
European (non-Finnish) 4 / 11,738 |
0.034% |
+ 5 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, European (Finnish), Middle Eastern)
ClinVar
This variant has been reported in ClinVar as Benign (6 clinical laboratories) and as Likely benign (4 clinical laboratories) and as benign (1 clinical laboratory). (ClinVarID = 353201)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.392. BayesDel score = 0.00236504.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. SDHA, a subunit of succinate dehydrogenase, is frequently altered by amplification in various cancers, including lung and bladder cancers.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53768747, n = 5 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 11 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
39321216 ↗
A Novel Human SDHA-Knockout Cell Line Model for the Functional Analysis of Clinically Relevant SDHA Variants.
CLINVAR
24893135 ↗
Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
33939658 ↗
The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma.
CLINVAR