NM_001001890.2:c.624C>T (p.Ala208=) is a synonymous variant in exon 4 of RUNX1 that does not alter the amino acid sequence. This variant is present at extremely low frequency in gnomAD (v4.1 grpmax FAF = 3.259e-05, 53/1,613,324 alleles), meeting PM2_Supporting under the MM-VCEP specification (MAF ≤0.00005).1 SpliceAI predicts no splice impact (max delta = 0.02), meeting BP4 (SpliceAI ≤0.20 for synonymous variants) under the MM-VCEP specification.2 The variant meets BP7 as a synonymous change not located in the critical splice region (not in the last 3 nucleotides of exon 4 nor the first nucleotide after the acceptor site), with SpliceAI ≤0.20.3 The ClinGen Myeloid Malignancy VCEP classified this variant as Likely Benign (ClinVar ID 532686, SCV001366061, reviewed by expert panel), consistent with the computed Tavtigian score of -1 (PM2_Supporting +1, BP4 -1, BP7 -1).4 No pathogenic evidence was identified: no de novo occurrences (PS2/PM6), no probands with FPD/AML phenotype (PS4), no functional data demonstrating damaging effect (PS3), no cosegregation data (PP1), the variant lies outside the RHD (PM1 not met), and in silico predictions do not support pathogenicity (PP3 not met).5 Population frequencies are too low to meet BA1 (>0.15%) or BS1 (0.015%-0.15%), but the variant is not absent from population databases.6 BS3 could not be independently assessed as the primary functional assay data reviewed by the expert panel is not publicly accessible through ClinVar; however, the expert panel's Likely Benign classification implies functional evidence was not supportive of pathogenicity.7
RUNX1
Final classification
VUS
RUNX1 c.624C>T · p.Ala208=
RUNX1
NM_001001890.2:c.624C>T (p.Ala208=) is a synonymous variant in exon 4 of RUNX1 that does not alter the amino acid sequence.
Tavtigian et.al., 2020 - Bayesian adaptation of Richards et.al., 2015 v3.1.0 point-based framework: PM2 supporting (+1) = 1 points, which maps to VUS.
Classification rationale
PM2
BP4BP6BP7
VUS
RUNX1 c.624C>T
PM2 + BP4 + BP6 + BP7
→
VUS
Gene diagram
· NM_001001890.2 · variants mapped to exon structure
RUNX1
NM_001001890.2
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 12 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
PM2_Supporting is met under the MM-VCEP specification. The grpmax filtering allele frequency (FAF) in gnomAD v4.1 is 3.259e-05 (0.00326%), which is below the VCEP threshold of ≤0.00005 (0.005%). The variant is present in ≥2,000 alleles tested (1,613,324 total alleles).
gnomAD v4.1 grpmax FAF = 3.259e-05 ≤ 0.000051613
✓
BP4
supporting
Benign
BP4 is met under the MM-VCEP specification for synonymous variants. The VCEP rule for synonymous variants requires SpliceAI ≤0.20. SpliceAI predicts no significant splice impact (max delta score = 0.02), which is well below the 0.20 threshold. Multiple lines of computational evidence suggest no impact on splicing.
SpliceAI max delta = 0.02 ≤ 0.20 (BP4 threshold for synonymous variants)
✓
✓
BP7
supporting
Benign
BP7 is met under the MM-VCEP specification. The variant is a synonymous change (c.624C>T, p.Ala208=) located in exon 4, not within the last 3 nucleotides preceding the canonical donor splice site (c.722-724) or the first nucleotide following the canonical acceptor splice site (c.533). SpliceAI predicts no splice impact (max delta = 0.02 ≤ 0.20). Conservation data is no longer required per the VCEP v3.1.0 update.
Synonymous variant not in critical splice region (not in last 3 nt of exon 4not first nt after acceptor)SpliceAI max delta = 0.02 ≤ 0.20
Assessed · not applied
Pathogenic
PS2
No proven de novo occurrences (with both maternity and paternity confirmed) of c.624C>T have been reported in patients with FPD/AML phenotype.
PS3
No well-established functional studies demonstrate a damaging effect for this variant.
PS4
PS4 requires ≥4 probands meeting RUNX1-phenotypic criteria (OR 127.1) for Strong, 2-3 probands for Moderate, or 1 proband for Supporting.
PM1
PM1 requires the variant to affect a residue within the Runt homology domain (RHD, AA 89-204).
PM6
PM6 requires ≥2 assumed de novo occurrences (PM6_Supporting) or ≥4 (PM6) in patients with FPD/AML phenotype, without confirmation of maternity and paternity.
PP1
PP1 requires ≥3 meioses (Supporting), 5-6 meioses (Moderate), or ≥7 meioses (Strong) of cosegregation with disease in affected families.
PP3
PP3 for synonymous variants under the MM-VCEP requires SpliceAI ≥0.38, including the creation of cryptic novel splice sites.
Benign
BA1
BA1 under the MM-VCEP specification requires a minor allele frequency ≥0.0015 (0.15%) in any general continental population dataset with ≥2,000 alleles tested and ≥5 variant alleles.
BS1
BS1 under the MM-VCEP specification requires a minor allele frequency between 0.00015 (0.015%) and 0.0015 (0.15%) in any general continental population dataset with ≥2,000 alleles tested and ≥5 variant alleles.
BS3
BS3 requires well-established functional studies (transactivation assays) showing no damaging effect.
BS4
BS4 under the MM-VCEP specification requires observation in ≥2 informative meioses (genotype-negative, phenotype-positive family members).
BP2
BP2 requires observation of the variant in trans with a known pathogenic variant in an individual without FPD/AML phenotype, or in cis with a pathogenic variant.
N/A · 9
PVS1 · PS1 · PM5 · PP2 · PP4 · PP5 · BS2 · BP1 · BP5
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
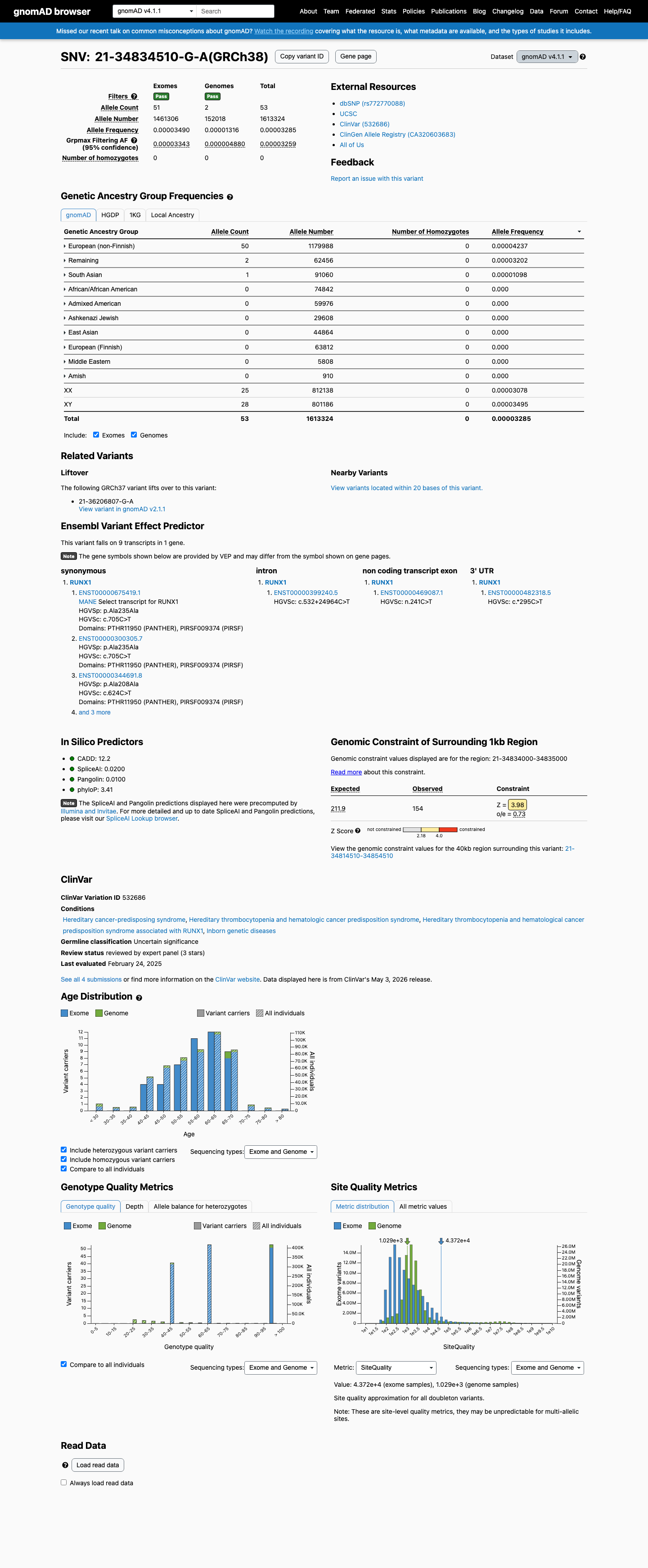
This variant is present in gnomAD v4.1 (AF= 3.28514e-05; MAF= 0.00329%, 53/1613324 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 4.23733e-05; MAF= 0.00424%, 50/1179988 alleles, homozygotes = 0); grpmax FAF= 3.259e-05.
v2.1
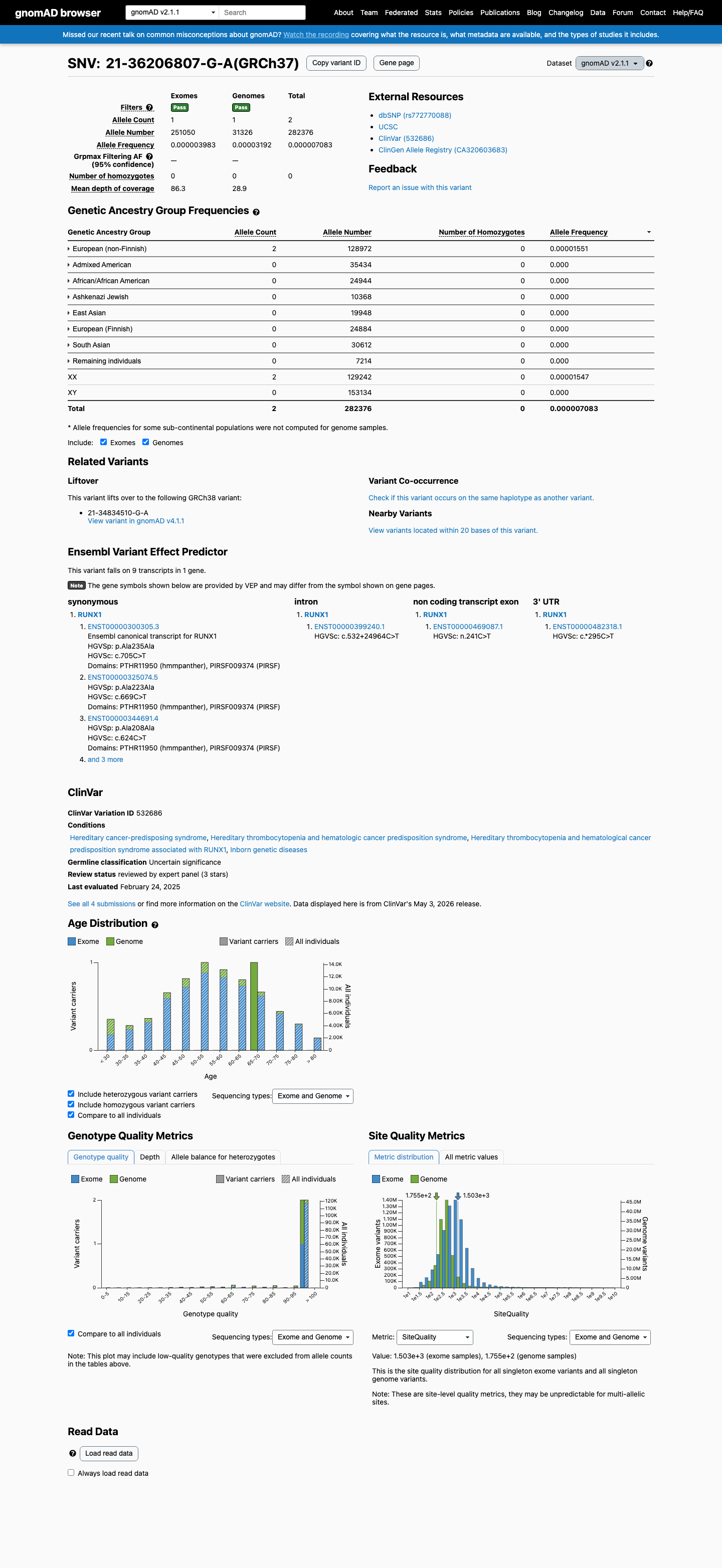
This variant is present in gnomAD v2.1 (AF= 7.08275e-06; MAF= 0.00071%, 2/282376 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.55072e-05; MAF= 0.00155%, 2/128972 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0033%
· 53 / 1,613,324
0 hom · FAF 0.0033%
0 hom · FAF 0.0033%
European (non-Finnish) 50 / 1,179,988 |
0.0042% |
Remaining individuals 2 / 62,456 |
0.0032% |
South Asian 1 / 91,060 |
0.0011% |
+ 7 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.00071%
· 2 / 282,376
0 hom
0 hom
European (non-Finnish) 2 / 128,972 |
0.0016% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
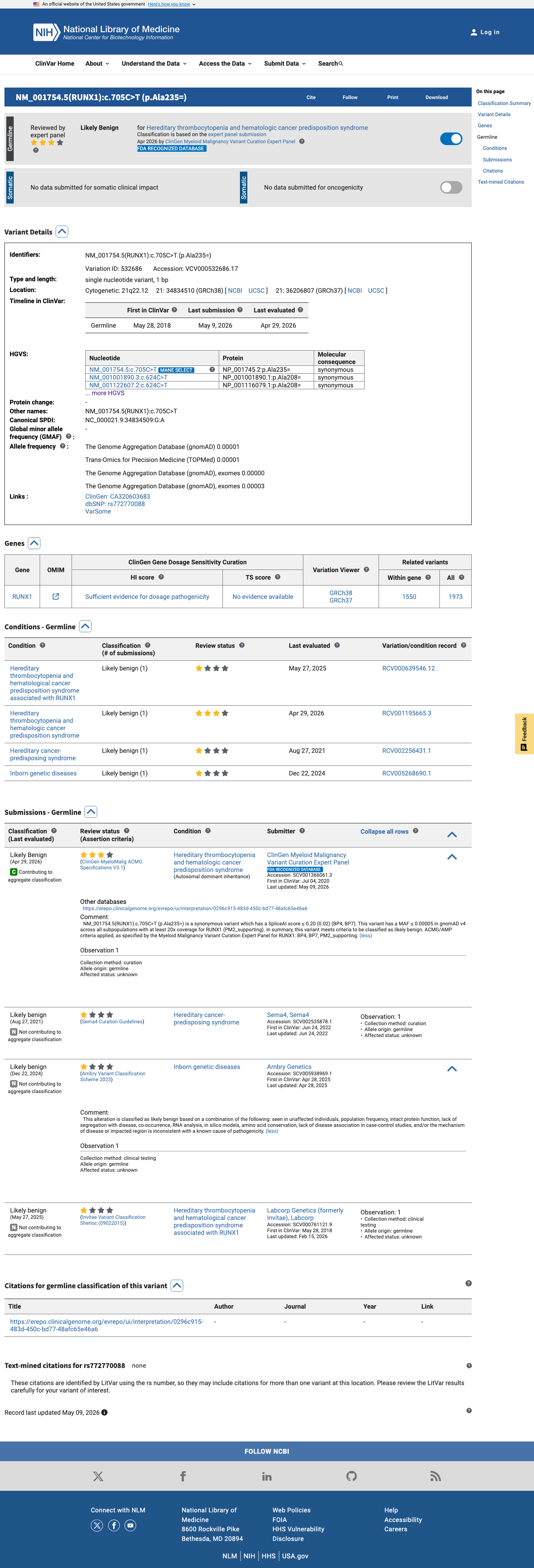
This variant has been reported in ClinVar as Likely benign (2 clinical laboratories) and as Likely Benign by ClinGen Myeloid Malignancy Variant Curation Expert Panel (expert panel). (ClinVarID = 532686)
In silico
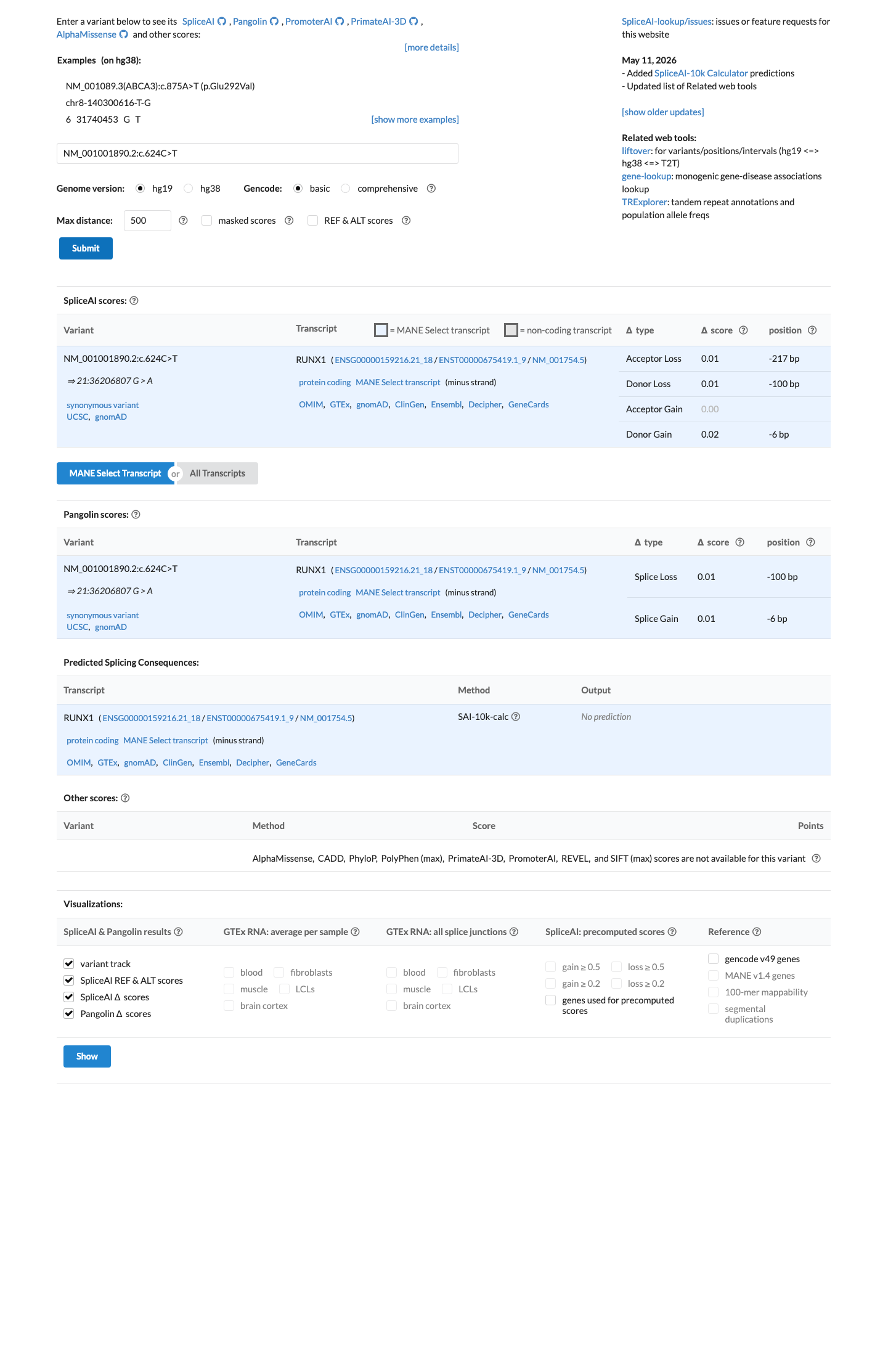
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 14 PMIDs not cited in assessment
23652378 ↗
A framework to start the debate on neonatal screening policies in the EU: an Expert Opinion Document.
CLINVAR
25626707 ↗
Whole-genome sequencing in newborn screening? A statement on the continued importance of targeted approaches in newborn screening programmes.
CLINVAR
25730230 ↗
Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine.
CLINVAR
23169492 ↗
The perspective from EASAC and FEAM on direct-to-consumer genetic testing for health-related purposes.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
22947299 ↗
Specific guidelines for assessing and improving the methodological quality of economic evaluations of newborn screening.
CLINVAR
23037933 ↗
Including the initial newborn screening bloodspot collection device serial number on birth certificates: basis and recommendations from the Secretary of Health and Human Services' Advisory Committee on Heritable Disorders in Newborns and Children.
CLINVAR
23881473 ↗
Newborn screening: education, consent, and the residual blood spot. The position of the national society of genetic counselors.
CLINVAR
24022298 ↗
Offering prenatal diagnostic tests: European guidelines for clinical practice [corrected].
CLINVAR
24394680 ↗
Parental permission for pilot newborn screening research: guidelines from the NBSTRN.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR