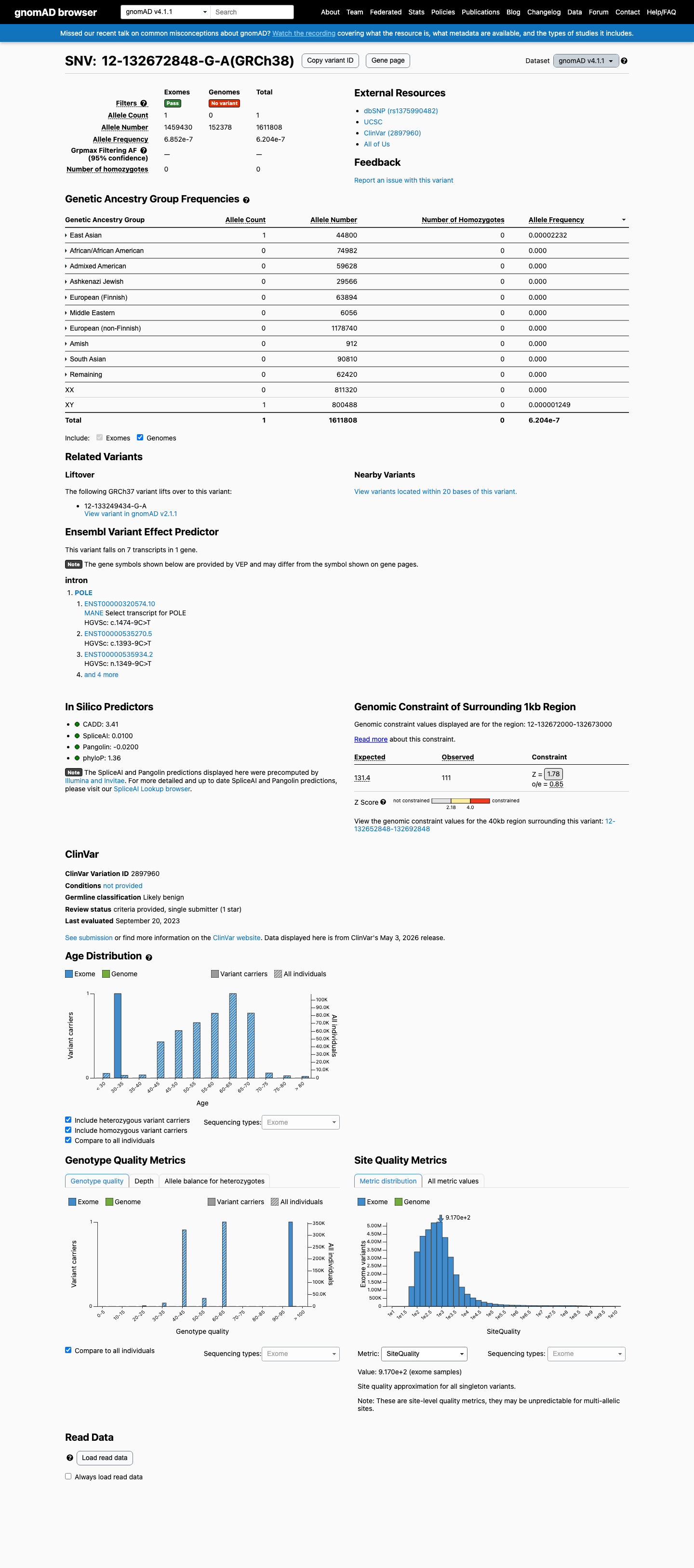
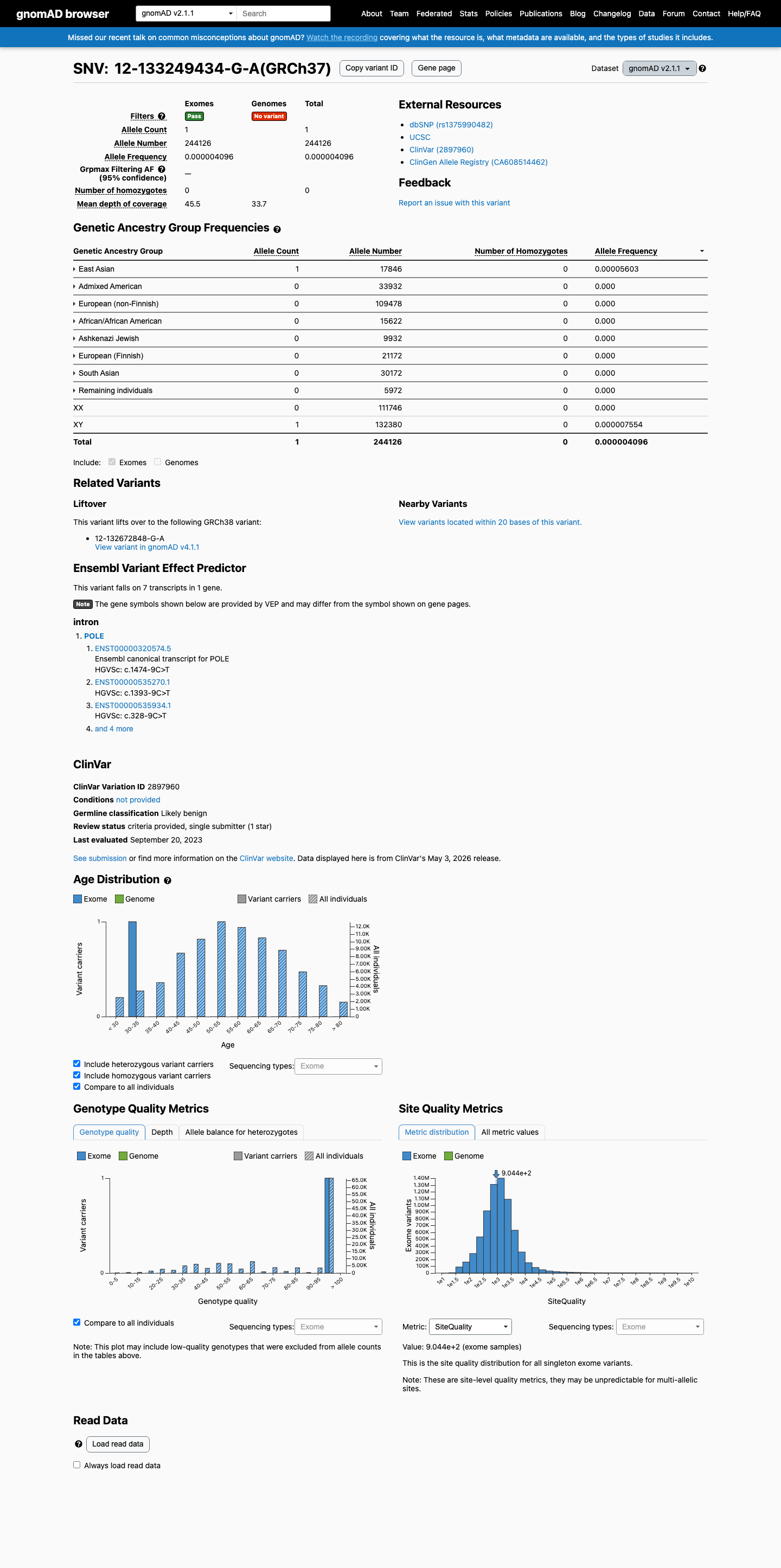
NM_006231.4:c.1474-9C>T is an intronic substitution in POLE located 9 bases upstream of exon 14. SpliceAI predicts no significant splicing impact (max delta score = 0.01), indicating the variant is unlikely to alter normal splicing.1 This variant is extremely rare in population databases, with an allele frequency of 4.10e-06 in gnomAD v2.1 (1/244,126 alleles) and 6.20e-07 in gnomAD v4.1 (1/1,611,808 alleles), both meeting the PM2_Supporting threshold for absence from population databases.2 ClinVar classifies this variant as Likely benign (VariationID 2897960, Labcorp Genetics, criteria provided, single submitter), supporting benign interpretation under BP6_Supporting.3 SpliceAI predicts no significant splice impact (max delta score = 0.01), consistent with a neutral computational assessment under BP4_Supporting.4 No pathogenic evidence criteria are met. The custom León-Castillo PM1 and PS4 rules are missense-specific and do not apply to this intronic substitution. The variant is absent from COSMIC and the León-Castillo supplementary tables. No de novo observations, functional studies, or segregation data are available.5 Applying the custom León-Castillo framework with generic ACMG/AMP 2015 fallback: the variant accrues one pathogenic supporting criterion (PM2_Supporting) and two benign supporting criteria (BP4_Supporting, BP6_Supporting). Under the likely_benign combination rules of the framework, two supporting benign criteria are sufficient for a classification of Likely benign. This classification is consistent with the existing ClinVar classification.6
POLE
Final classification
Likely Benign
POLE c.1474-9C>T · p.?
POLE
NM_006231.4:c.1474-9C>T is an intronic substitution in POLE located 9 bases upstream of exon 14. SpliceAI predicts no significant splicing impact (max delta score = 0.01), indicating the variant is unlikely to alter normal splicing.
The custom León-Castillo et al. 2020 POLE framework was used as the primary classification framework. Only supporting-strength criteria are met: one pathogenic (PM2_Supporting) and two benign (BP4_Supporting, BP6_Supporting). Under the framework's likely_benign combination rules, >=2 Supporting benign criteria are sufficient for a Likely Benign classification.
Classification rationale
PM2
BP4BP6
Likely Benign
POLE c.1474-9C>T
PM2 + BP4 + BP6
→
Likely Benign
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 16 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low allele frequency in population databases: gnomAD v2.1 AF = 4.10e-06 (1/244,126 alleles, 0 homozygotes) and gnomAD v4.1 AF = 6.20e-07 (1/1,611,808 alleles, 0 homozygotes). Both frequencies are well below the 0.1% PM2 threshold. The variant is absent from gnomAD-Canada v1.0.
gnomAD v2.1: 1/244126 alleles (AF=4.10e-06)0 homozygotes
✓
BP4
supporting
Benign
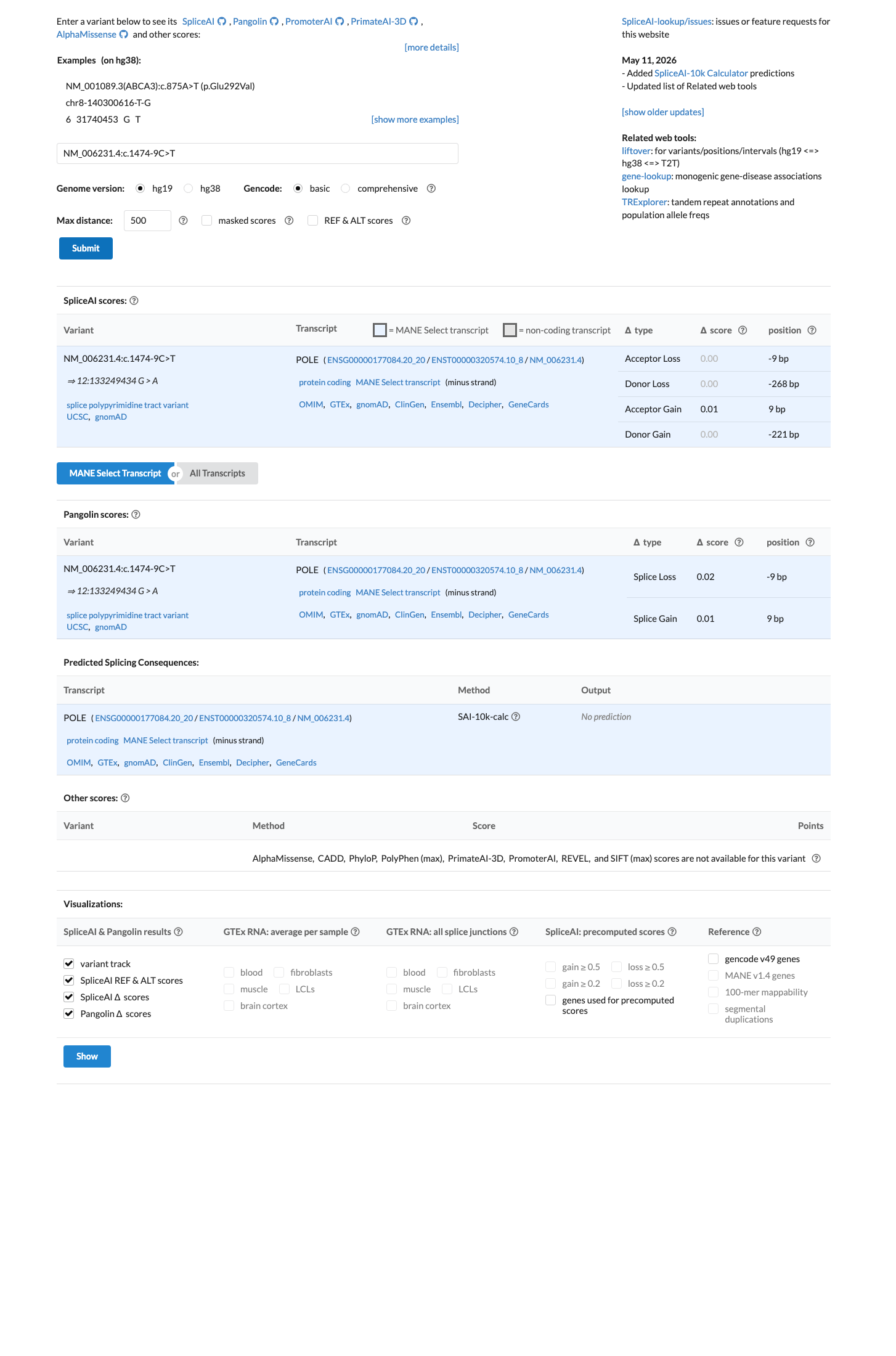
SpliceAI predicts no significant splice impact for this intronic substitution (max delta score = 0.01), suggesting the variant is unlikely to disrupt normal splicing of POLE. The custom León-Castillo BP4 rule applies only to missense variants in Supplementary Tables S2/S3 and is not triggered here; under generic BP4 assessment, computational evidence supports a neutral effect.
SpliceAI max delta = 0.01 (well below commonly used thresholds of 0.1-0.2 for splice effect prediction)REVELBayesDel not applicable to intronic variants
✓
BP6
supporting
Benign
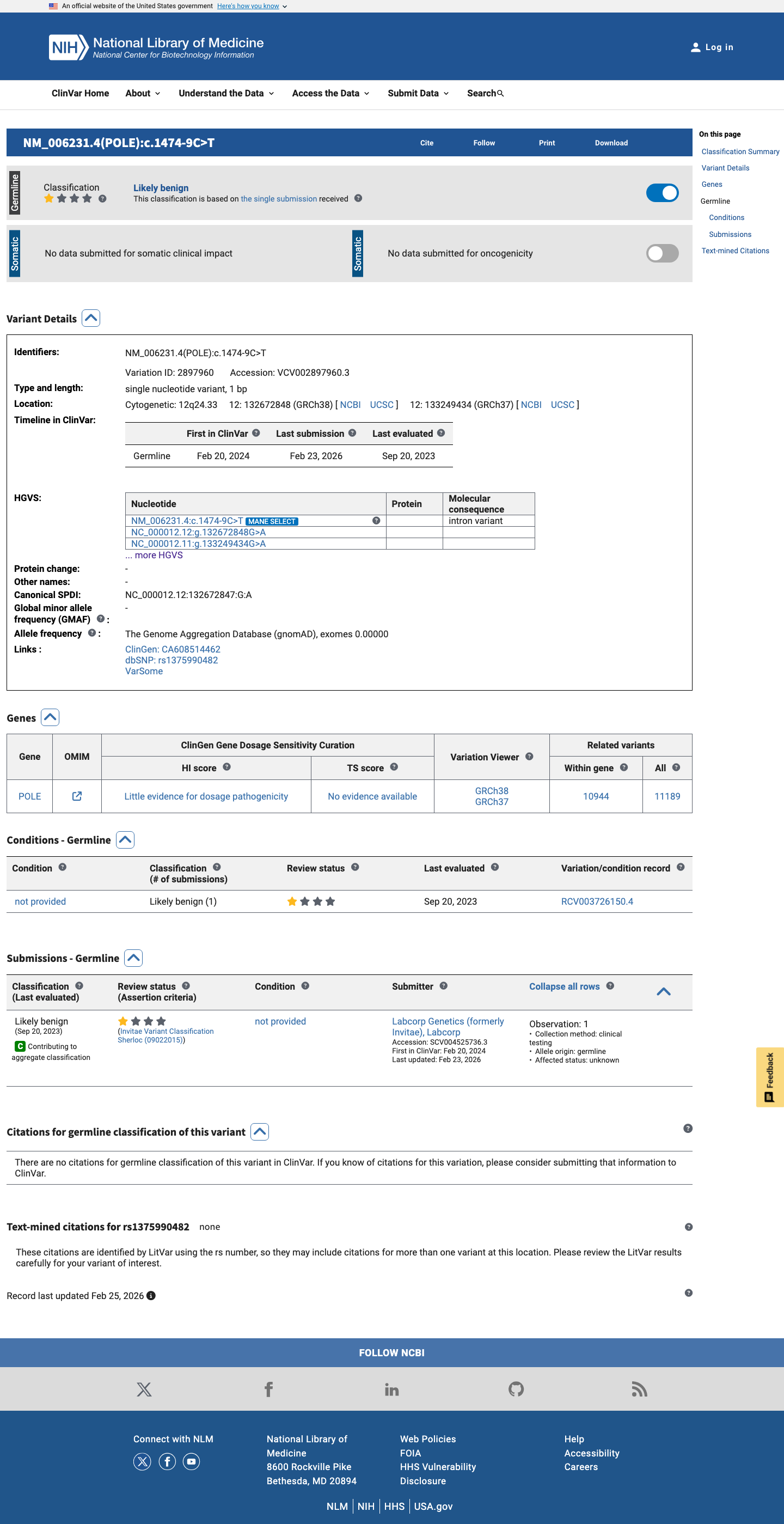
This variant has been classified as Likely benign by a clinical diagnostic laboratory (Labcorp Genetics, formerly Invitae; ClinVar SCV004525736, VariationID 2897960) with criteria provided. The submitter is a reputable clinical testing laboratory.
ClinVar Likely benign classification from Labcorp Genetics (SCV004525736)Criteria providedsingle submitter
Assessed · not applied
Pathogenic
PS2
No de novo observation of NM_006231.4:c.1474-9C>T has been reported in the literature or clinical databases.
PS3
No functional studies (e.g., RNA splicing assays, minigene constructs, polymerase activity assays) have been identified that evaluate the functional impact of NM_006231.4:c.1474-9C>T.
PS4
The custom León-Castillo PS4 rule applies only to missense variants recurrent in COSMIC and TCGA endometrial carcinoma cohorts with combined EC count ≥10.
PM1
The custom León-Castillo PM1 rules apply only to specific missense variants in the exonuclease domain (five established hotspots at PM1_Strong, specific recurrent variants at PM1_Moderate, and specific uncertain-tier variants at PM1_Supporting).
PM6
No de novo observation of NM_006231.4:c.1474-9C>T has been reported in the literature or clinical databases.
PP1
No co-segregation data are available for NM_006231.4:c.1474-9C>T.
PP3
The custom León-Castillo PP3 rule applies only to missense variants present in Supplementary Tables S2 or S3 with REVEL class 'likely disease causing' and ≤1 benign in silico result.
PP4
No phenotype or clinical data specifically associated with NM_006231.4:c.1474-9C>T have been identified.
PP5
ClinVar classifies this variant as Likely benign (VariationID 2897960, Labcorp Genetics, criteria provided, single submitter).
Benign
BA1
The allele frequency in gnomAD v2.1 (AF = 4.10e-06, 0.0004%) and v4.1 (AF = 6.20e-07, 0.00006%) is far below the 1% BA1 threshold.
BS1
The allele frequency in gnomAD v2.1 (AF = 4.10e-06, 0.0004%) is far below the 0.3% BS1 threshold.
BS2
The variant is not observed in a homozygous state in gnomAD (0 homozygotes in both v2.1 and v4.1).
BS3
No functional studies (e.g., RNA splicing assays, minigene constructs, enzymatic activity assays) assessing the impact of NM_006231.4:c.1474-9C>T have been identified.
BS4
No segregation data are available for NM_006231.4:c.1474-9C>T.
BP2
No observations of NM_006231.4:c.1474-9C>T in trans with a known pathogenic POLE variant have been reported.
BP5
No observations of NM_006231.4:c.1474-9C>T in cases with an alternate molecular cause for disease have been identified.
N/A · 9
PVS1 · PS1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.20421e-07; MAF= 0.00006%, 1/1611808 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 2.23214e-05; MAF= 0.00223%, 1/44800 alleles, homozygotes = 0).
v2.1
This variant is present in gnomAD v2.1 (AF= 4.09625e-06; MAF= 0.00041%, 1/244126 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 5.6035e-05; MAF= 0.00560%, 1/17846 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,611,808
0 hom
0 hom
East Asian 1 / 44,800 |
0.0022% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.00041%
· 1 / 244,126
0 hom
0 hom
East Asian 1 / 17,846 |
0.0056% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 2897960)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR