NM_001202543.1:c.2983C>T (p.Arg995Ter) is a nonsense variant in exon 19 of CUX1 predicted to undergo nonsense-mediated decay, meeting PVS1 at very strong strength.1 The variant is absent from gnomAD v2.1 (0/251,110 alleles) and v4.1, meeting PM2 at supporting strength.2 The variant has been classified as Pathogenic in ClinVar (Variation ID: 4532047) by a reputable clinical diagnostic laboratory (Victorian Clinical Genetics Services), meeting PP5 at supporting strength.3 Under ACMG/AMP 2015 combination rules, one Very Strong criterion (PVS1) plus two Supporting criteria (PM2, PP5) is sufficient for a Pathogenic classification.4 Heterozygous loss-of-function variants in CUX1 cause an autosomal dominant neurodevelopmental disorder characterized by global developmental delay, intellectual disability, hypotonia, and seizures, as established by Oppermann et al. (2023) in a cohort of 34 individuals.5 No benign evidence was identified: the variant is too rare for BA1/BS1, no functional studies demonstrate a benign effect, and no lack of segregation or alternate molecular basis has been reported.6 Final classification: Pathogenic. PVS1_VeryStrong + PM2_Supporting + PP5_Supporting.7
CUX1
Final classification
Pathogenic
CUX1 c.2983C>T · p.Arg995Ter
CUX1
NM_001202543.1:c.2983C>T (p.Arg995Ter) is a nonsense variant in exon 19 of CUX1 predicted to undergo nonsense-mediated decay, meeting PVS1 at very strong strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 supporting, PP5 supporting; combination = 1 very strong + 2 supporting, which maps to Pathogenic.
Classification rationale
PVS1PM2PP5
Pathogenic
CUX1 c.2983C>T
PVS1 + PM2 + PP5
→
Pathogenic
Gene diagram
· NM_001202543.1 · variants mapped to exon structure
CUX1
NM_001202543.1
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 19 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_001202543.1:c.2983C>T introduces a premature termination codon (p.Arg995Ter) at codon 995 of 1517 in exon 19 of 24. The premature stop is located >55 nt upstream of the last exon-exon junction (c.3921), predicting nonsense-mediated decay. Heterozygous loss-of-function variants in CUX1 are an established disease mechanism for autosomal dominant CUX1-related neurodevelopmental disorder, characterized by developmental delay, intellectual disability, hypotonia, and seizures (Oppermann et al., 2023, PMID:37644171). Under ClinGen SVI PVS1 recommendations (PMC6185798), this qualifies for PVS1 at very strong strength.
Nonsense variant creating premature stop codon (p.Arg995Ter) at amino acid 995 of 1517NMD predicted: PTC located at c.2983 in exon 19>55 nt upstream of last exon-exon junction at c.3921
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD v2.1 (0/251,110 alleles, allele frequency 0.0%) across all populations. The variant was also not detected in gnomAD v4.1, consistent with extreme rarity in the general population. This supports a pathogenic interpretation under PM2 (absent from population controls).
Absent from gnomAD v2.1: 0/251110 allelesAF=0.0%
✓
PP5
supporting
Pathogenic
This variant has been classified as Pathogenic in ClinVar (Variation ID: 4532047) by Victorian Clinical Genetics Services, Murdoch Childrens Research Institute, a reputable clinical diagnostic laboratory (SCV007108443, criteria provided, clinical testing). The submitter cites PMID:37644171 (Oppermann et al., 2023) and PMID:25741868 (ACMG guidelines). While the general gene-level evidence is accessible, the specific clinical case details underlying the submission (individual phenotype, segregation, detailed criteria application) are not fully available for independent evaluation, satisfying the PP5 requirement that evidence from the reputable source is not fully available to the interpreting laboratory.
ClinVar Variation ID 4532047: Pathogeniccriteria providedsingle submitter
Assessed · not applied
Pathogenic
PS2
No de novo observation with confirmed maternity and paternity has been identified for this specific variant.
PS3
No functional studies directly assessing the biological effect of NM_001202543.1:c.2983C>T were identified.
PS4
The variant has been observed in a cohort of individuals with CUX1-related neurodevelopmental disorder (Oppermann et al., 2023), but no case-control study demonstrating statistically significant enrichment in affected individuals versus population controls is available.
PM1
The variant does not lie in a statistically significant mutational hotspot (Cancer Hotspots analysis: residue not significant, exact variant not listed).
PM6
No publication reports this variant as assumed de novo (without confirmation of maternity/paternity).
PP1
No cosegregation data are available.
PP2
PP2 applies to missense variants in genes where missense variation is a known disease mechanism and benign missense variation is rare.
PP3
In silico tools do not support a deleterious effect at the nucleotide level.
PP4
Insufficient phenotype specificity data are available for this specific variant.
Benign
BA1
This variant has an allele frequency of 0.0% in gnomAD v2.1 (0/251,110 alleles), well below the BA1 threshold of >1.0%.
BS1
This variant has an allele frequency of 0.0% in gnomAD v2.1, well below the BS1 threshold of >0.3%.
BS2
The variant has not been observed in a homozygous state in population controls.
BS3
No functional studies demonstrating a benign effect for this specific variant were identified.
BS4
No evidence of lack of segregation in affected family members.
BP1
BP1 applies to missense variants in genes where a truncating mechanism is the primary cause of disease.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on the gene product.
BP5
BP5 applies when a variant is found in a case with an alternate molecular basis for disease.
BP6
BP6 requires a reputable source to classify the variant as benign or likely benign.
BP7
BP7 applies to synonymous variants predicted to have no impact on splicing.
N/A · 6
PS1 · PM3 · PM4 · PM5 · BP2 · BP3
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
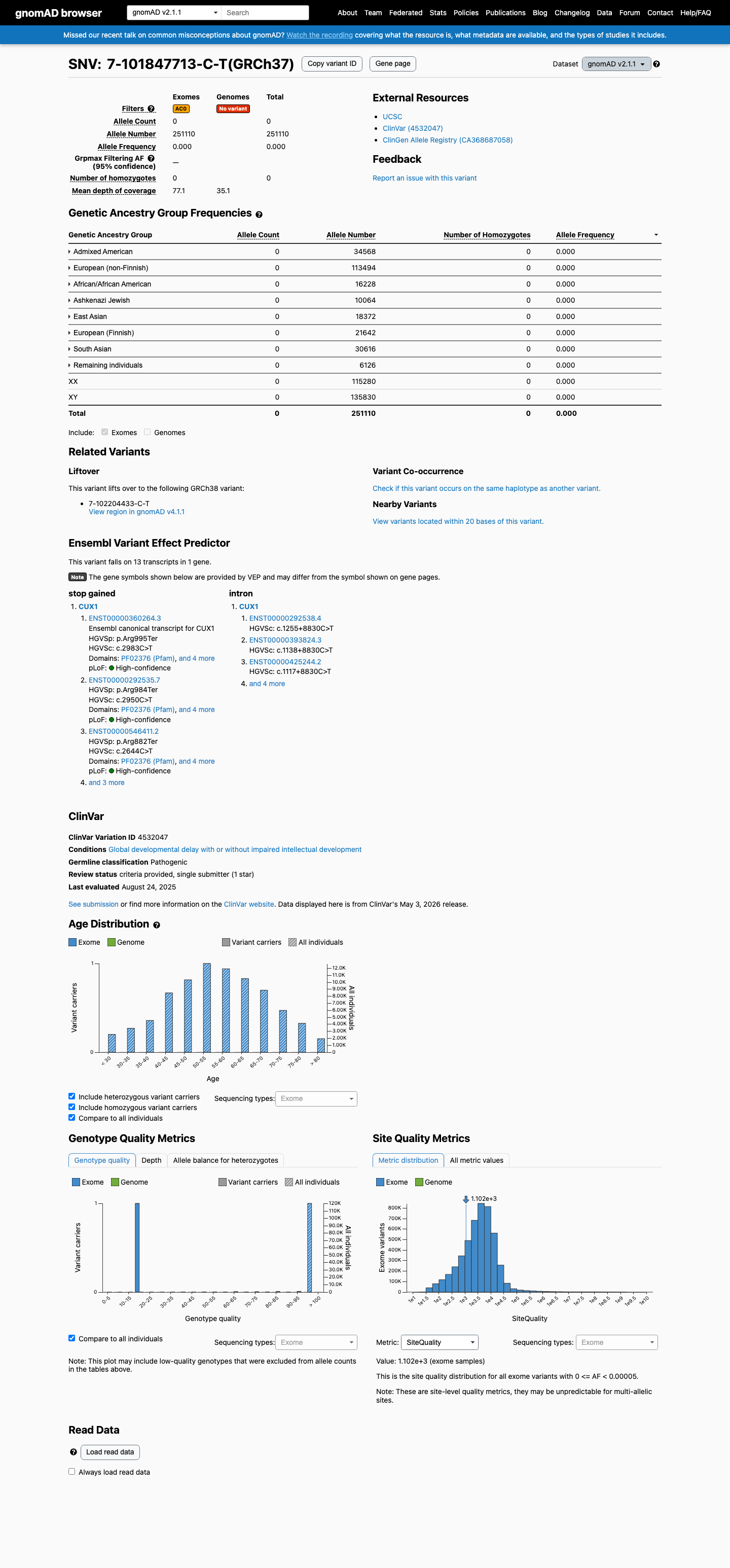
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/251110 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/16228 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / 251,110
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
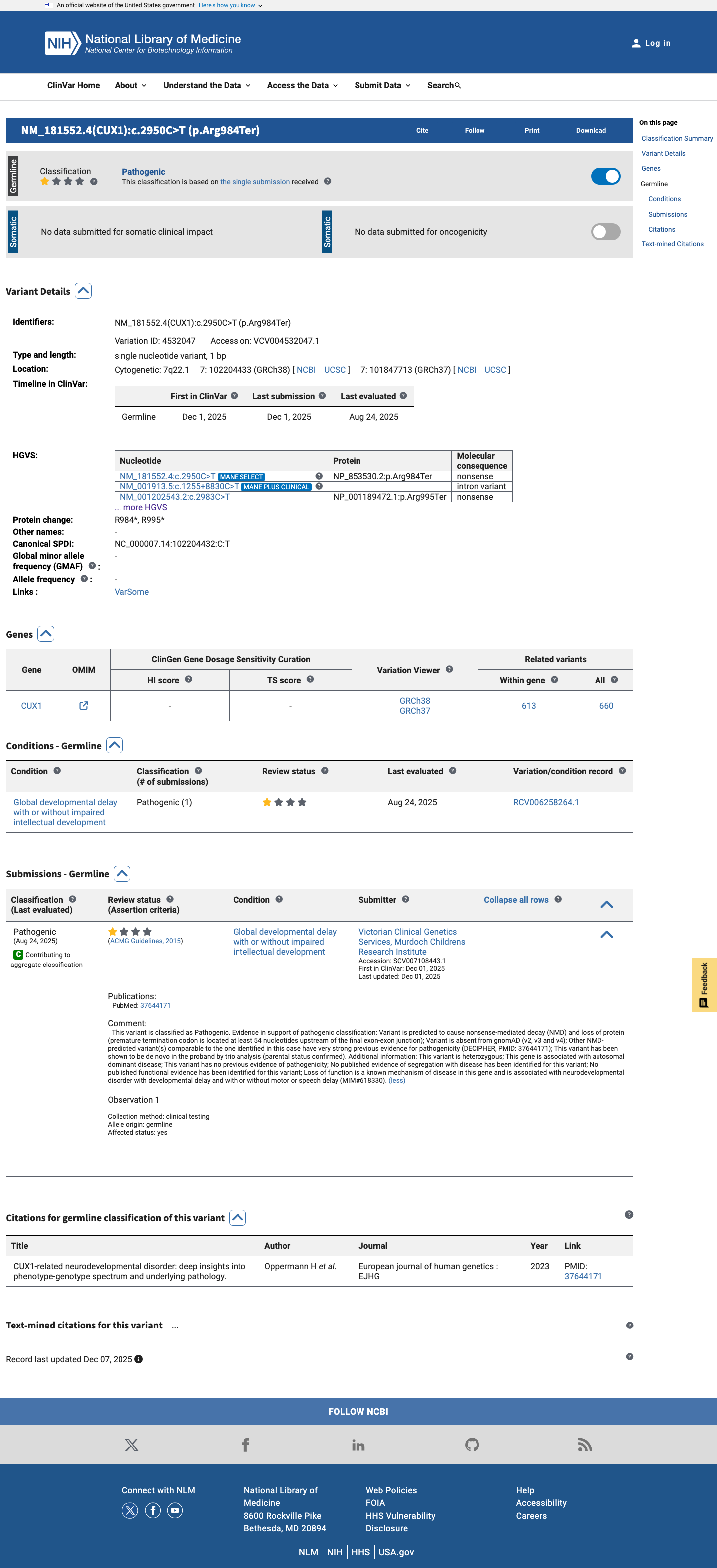
This variant has been reported in ClinVar as Pathogenic (1 clinical laboratory). (ClinVarID = 4532047)
In silico
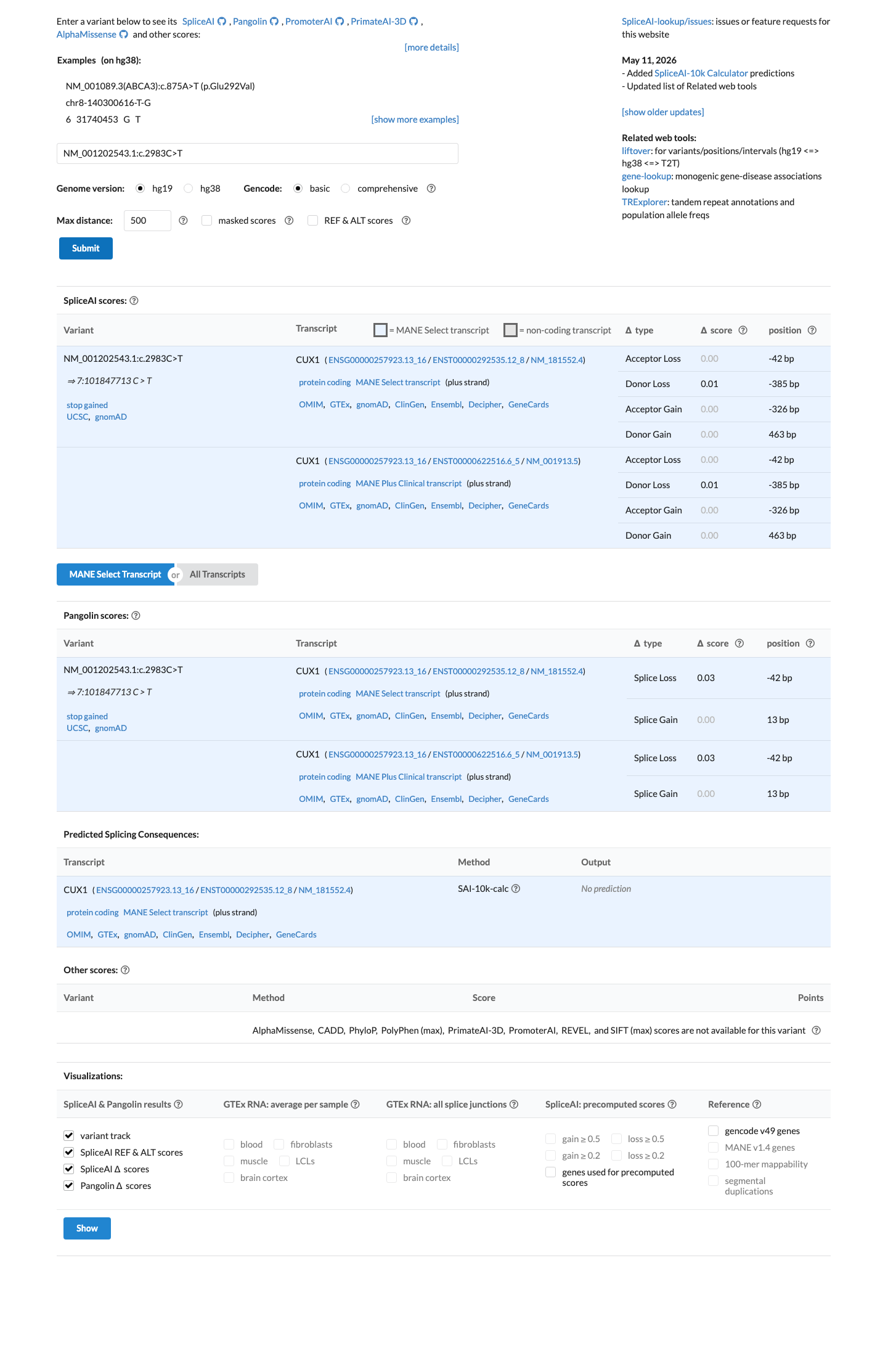
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). BayesDel score = 0.60222.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV107349027, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 3 further PMIDs triaged but not cited — see Sources & References.
CUX1-related neurodevelopmental disorder: deep insights into phenotype-genotype spectrum and underlying pathology.
Found
ClinVar Variation ID 4532047: Pathogenic criteria provided single submitter Submitter: Victorian Clinical Genetics Services Murdoch Childrens Research Institute (SCV007108443) Submission cites PMID:37644171 and PMID:25741868
Applied to
→PP5 supports · met
→PVS1 supports · met
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
23212519 ↗
CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia.
ONCOKB
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR