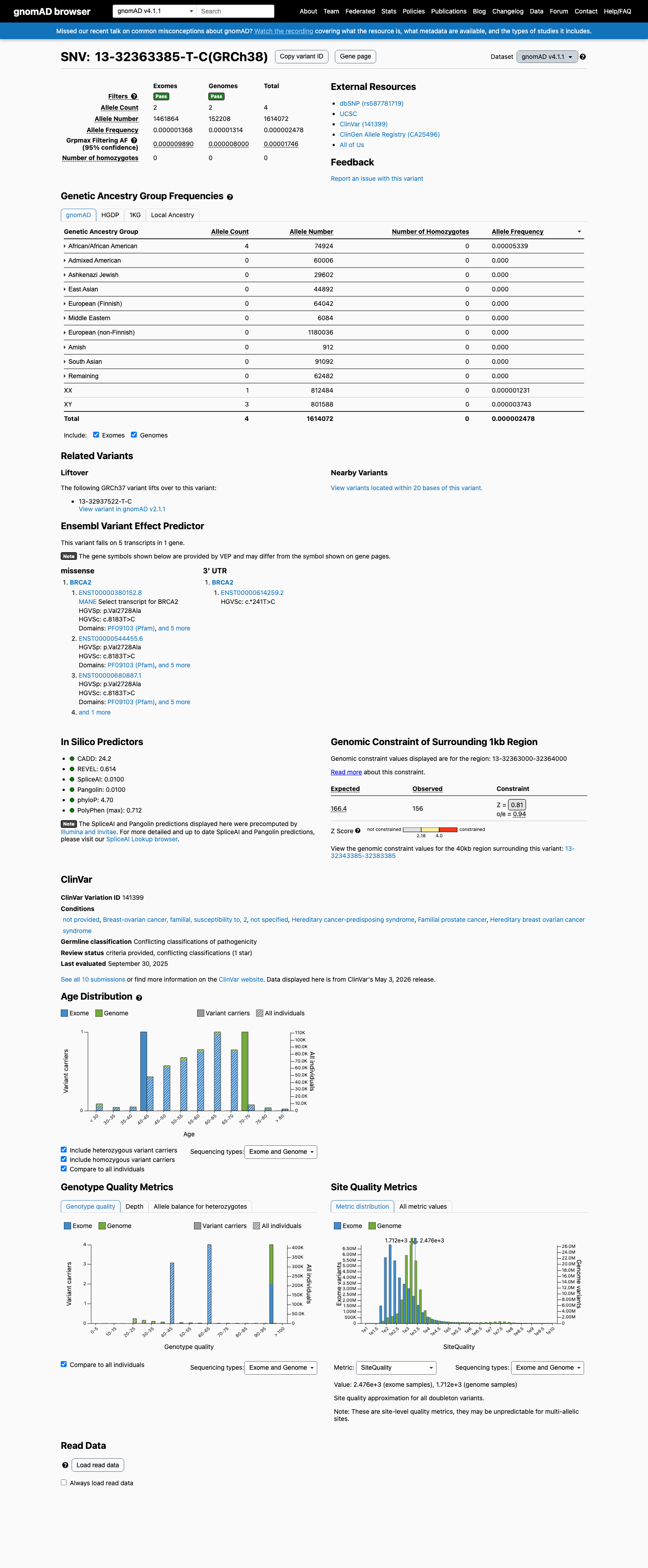
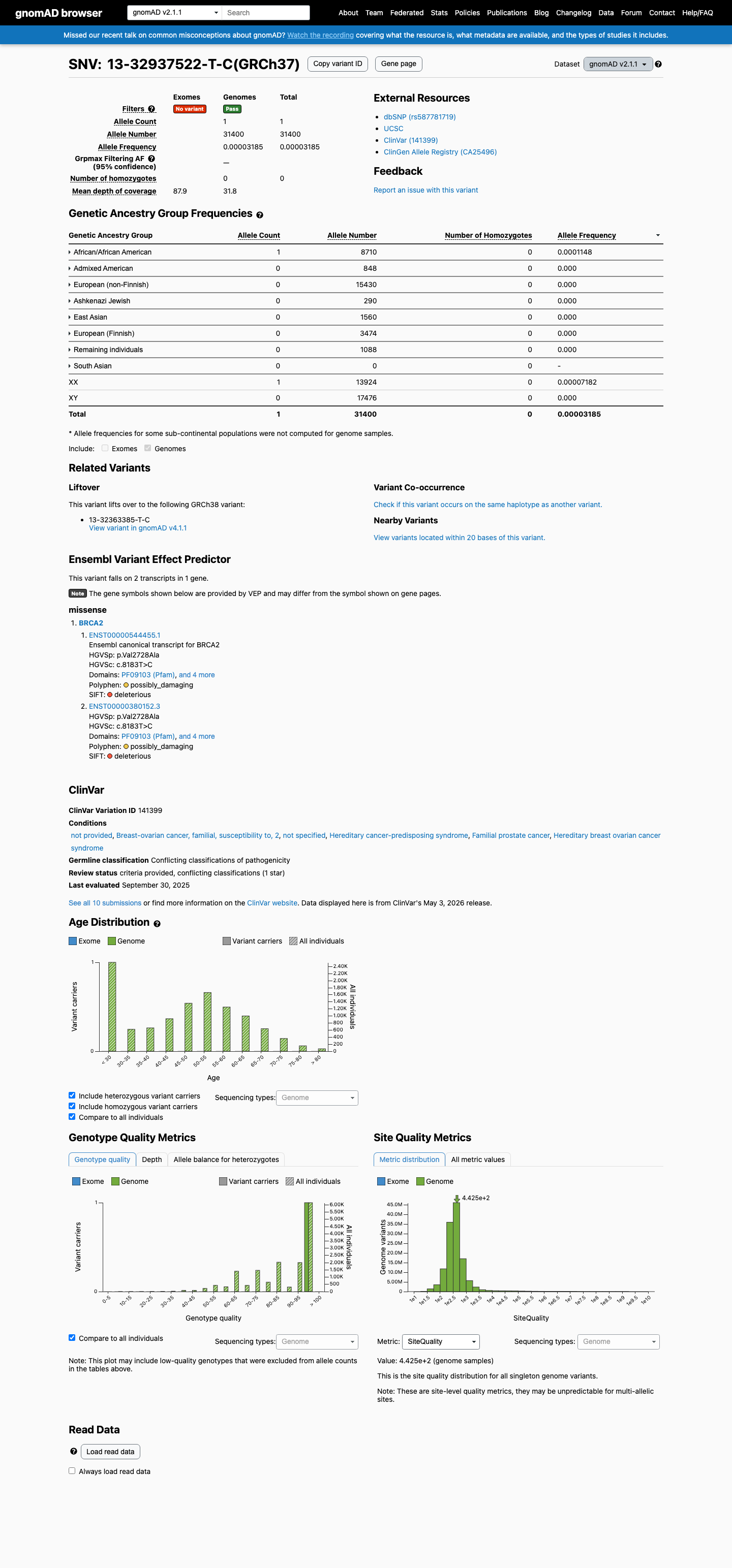
NM_000059.4:c.8183T>C (p.Val2728Ala) is a missense variant in exon 18 of BRCA2, located within the DNA binding domain (aa 2481-3186). This variant is present at extremely low frequency in population databases: gnomAD v2.1 (1/31,400 alleles, exclusively in genomes; exome not covered) and gnomAD v4.1 (4/1,614,072 alleles, grpmax FAF=1.746e-05). It is absent from gnomAD-Canada v1.0.1 In ClinVar (Variation ID 141399), this variant is classified as Uncertain Significance by seven clinical laboratories and Likely Benign by one laboratory (criteria provided, single submitter).2 ENIGMA Specifications Table 9 assigns BS3_Strong based on a calibrated functional study (Richardson et al. 2021, PMID:33609447) demonstrating that V2728A exhibits HDR activity similar to benign control variants.3 ENIGMA BP4_Supporting is met: the variant is inside the DNA binding domain with no predicted impact via protein change (BayesDel no-AF=0.103, ≤0.18) and no predicted splicing impact (SpliceAI max delta=0.01, ≤0.1).4 The clinical-history likelihood ratio from Li et al. 2020 (PMID:31853058) is LR=1.24 based on 1 proband, falling in the neutral zone and providing no evidence for PP4 or BP5.5 No pathogenic comparator exists at residue Val2728 (c.8182G>A p.Val2728Ile is classified as benign by ENIGMA), so PS1 is not met. PVS1, PM5, and other null-variant criteria are not applicable to this missense substitution.6 Applying ENIGMA Table 3 combination rules: BS3_Strong (1 Strong Benign) + BP4_Supporting (1 Supporting Benign) meets the likely benign rule [1 Strong (Benign) + 1 Supporting (Benign)]. The total benign evidence is one Strong and one Supporting criterion with no pathogenic criteria met.7
BRCA2
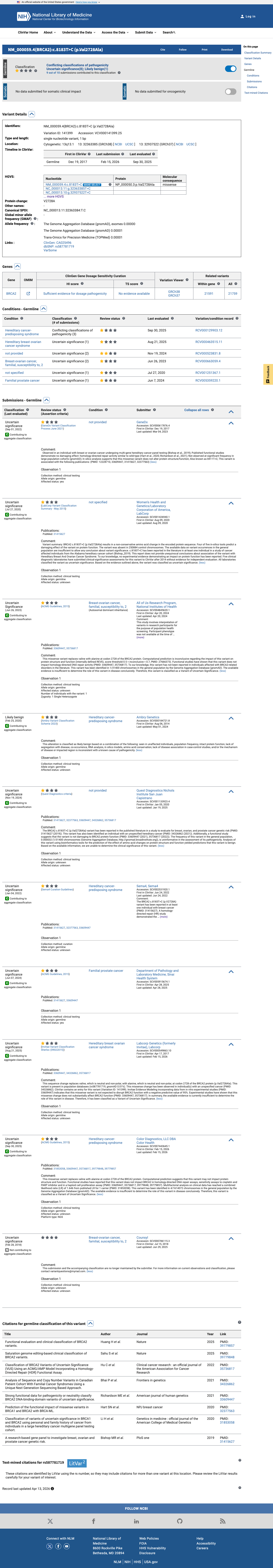
Final classification
Likely Benign
BRCA2 c.8183T>C · p.Val2728Ala
BRCA2
NM_000059.4:c.8183T>C (p.Val2728Ala) is a missense variant in exon 18 of BRCA2, located within the DNA binding domain (aa 2481-3186).
ENIGMA BRCA1/BRCA2 v1.2.0 Table 3 classification framework was used as the primary authority. The variant has 1 met Strong Benign criterion (BS3_Strong: calibrated functional assay shows HDR activity similar to benign controls, PMID:33609447) and 1 met Supporting Benign criterion (BP4_Supporting: BayesDel no-AF=0.103 ≤0.18, SpliceAI max delta=0.01 ≤0.1, inside DNA binding domain). No pathogenic criteria are met. The combination of 1 Strong (Benign) + 1 Supporting (Benign) satisfies the ENIGMA Table 3 Likely Benign rule. No conflicting evidence to resolve via the point system.
Classification rationale
BS3BP4
Likely Benign
BRCA2 c.8183T>C
BS3 + BP4
→
Likely Benign
3
vcep_specifications_table9_v1_2_2024_11_18PMID:33609447 ↗
4
bayesdelspliceai ↗cspec ↗
5
vcep_pmid_31853058_brca2_clinical_history_lrPMID:31853058 ↗
6
vcep_humu_40_1557_s001pm5_candidates
7
cspec ↗
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 12 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BS3
strong
Benign
ENIGMA Specifications Table 9 assigns BS3_Strong for c.8183T>C p.(Val2728Ala) based on a calibrated functional study (Richardson et al. 2021, PMID:33609447) demonstrating that this variant exhibits protein function similar to benign control variants in a homology-directed repair (HDR) assay.
ENIGMA Table 9 BS3_Strong assignment: 'Reported by one calibrated study to exhibit protein function similar to benign control variants (PMID:33609447) (BS3 met).'
✓
BP4
supporting
Benign
ENIGMA BP4_Supporting applies to missense variants inside a clinically important functional domain with no predicted impact via protein change or splicing: BayesDel no-AF score ≤0.18 AND SpliceAI ≤0.1. This variant satisfies both conditions (BayesDel=0.103, SpliceAI max delta=0.01) and is located within the BRCA2 DNA binding domain (aa 2481-3186).
BayesDel no-AF=0.103 (≤0.18)SpliceAI max delta=0.01 (≤0.1)residue 2728 is inside BRCA2 DNA binding domain (aa 2481-3186).
Assessed · not applied
Pathogenic
PS1
No previously classified pathogenic missense variant exists at the same amino acid residue (Val2728).
PS3
ENIGMA Specifications Table 9 assigns BS3 (not PS3) for this variant based on calibrated functional assay data showing protein function similar to benign control variants (PMID:33609447).
PS4
No case-control study demonstrates significantly increased prevalence of this variant in affected individuals versus controls.
PM2
ENIGMA PM2_Supporting requires absence from both gnomAD v2.1 non-cancer exome and v3.1 non-cancer.
PP1
No cosegregation data available for this variant.
PP3
ENIGMA PP3 requires BayesDel no-AF score ≥0.30 for missense variants inside a clinically important functional domain (or SpliceAI ≥0.2 for predicted splicing).
PP4
The clinical-history likelihood ratio from Li et al.
Benign
BA1
ENIGMA BA1 requires FAF >0.1% (0.001) in gnomAD non-founder populations.
BS1
ENIGMA BS1 requires FAF >0.002% in gnomAD v2.1 non-cancer exome or v3.1 non-cancer.
BS2
No evidence of this variant observed in a homozygous state in individuals without Fanconi Anemia phenotype.
BS4
No quantitative lack-of-segregation evidence is available.
BP5
The clinical-history likelihood ratio from Li et al.
N/A · 14
PVS1 · PS2 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.4782e-06; MAF= 0.00025%, 4/1614072 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 5.33874e-05; MAF= 0.00534%, 4/74924 alleles, homozygotes = 0); grpmax FAF= 1.746e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.18471e-05; MAF= 0.00318%, 1/31400 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0.000114811; MAF= 0.01148%, 1/8710 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,614,072
0 hom · FAF 0.0017%
0 hom · FAF 0.0017%
African/African American 4 / 74,924 |
0.0053% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, European (non-Finnish))
gnomAD v2.1
0.0032%
· 1 / 31,400
0 hom
0 hom
African/African American 1 / 8,710 |
0.011% |
+ 7 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (7 clinical laboratories) and as Uncertain Significance (1 clinical laboratory) and as Likely benign (1 clinical laboratory). (ClinVarID = 141399)
In silico
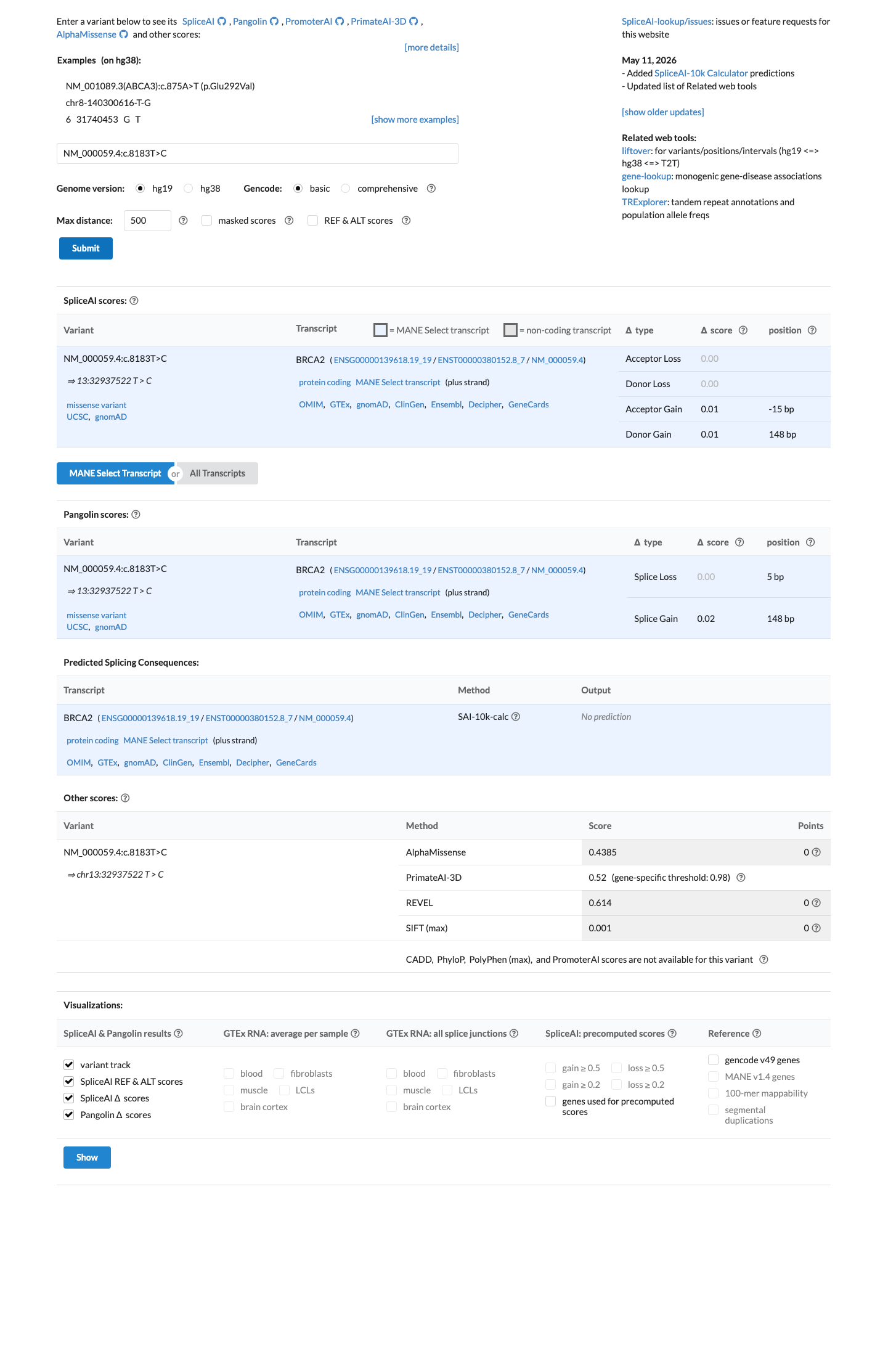
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.614. BayesDel score = 0.103001.
Functional
Likely Neutral
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Neutral; curated oncogenicity label: Likely Neutral.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV105932213, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance.
Found
ENIGMA Table 9 BS3_Strong assignment: 'Reported by one calibrated study to exhibit protein function similar to benign control variants (PMID:33609447) (BS3 met).'
Applied to
→BS3 supports · met
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
35736817 ↗
Classification of BRCA2 Variants of Uncertain Significance (VUS) Using an ACMG/AMP Model Incorporating a Homology-Directed Repair (HDR) Functional Assay.
ONCOKB
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
31415627 ↗
A research-based gene panel to investigate breast, ovarian and prostate cancer genetic risk.
CLINVAR
31853058 ↗
Classification of variants of uncertain significance in BRCA1 and BRCA2 using personal and family history of cancer from individuals in a large hereditary cancer multigene panel testing cohort.
CLINVAR
32377563 ↗
Prediction of the functional impact of missense variants in BRCA1 and BRCA2 with BRCA-ML.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR