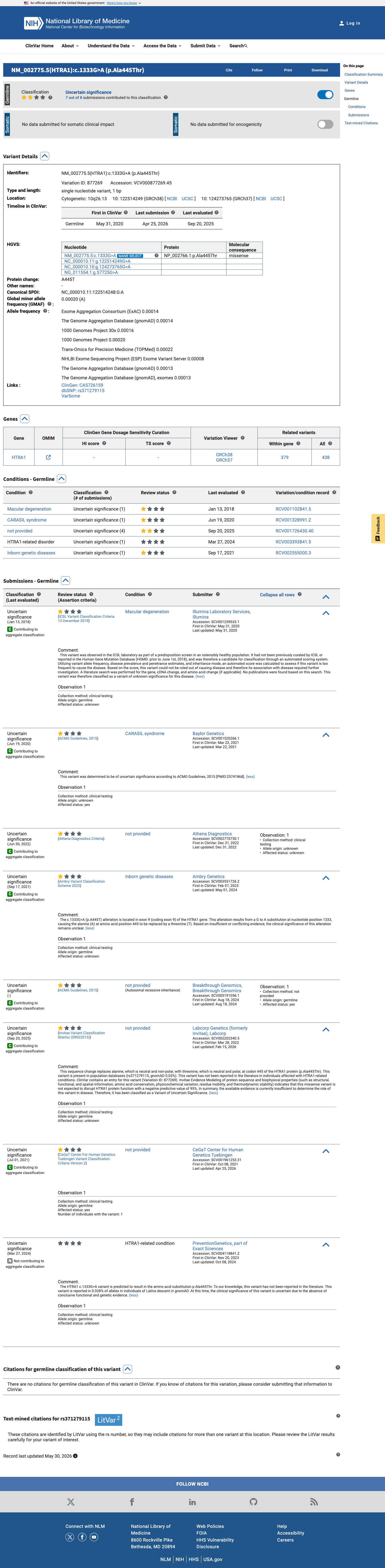
NM_002775.4:c.1333G>A (p.Ala445Thr) is a missense variant in HTRA1 located within the peptidase S1 serine protease domain (residues 157–473), a well-established functional domain critical for HTRA1 protease activity. Loss-of-function missense variants in this domain are a known cause of cerebral small vessel disease including CARASIL and CADASIL2.1 This variant is present at very low frequency in population databases: gnomAD v2.1 allele frequency is 0.012% (35/282,868 alleles, 0 homozygotes) and gnomAD v4.1 allele frequency is 0.010% (168/1,613,814 alleles, 0 homozygotes). The variant is absent from gnomAD-Canada. All frequencies fall well below the 0.1% PM2 threshold.2 Multiple in silico predictors suggest a neutral effect: REVEL score is 0.133 (below pathogenic threshold), BayesDel score is −0.219 (negative, favoring benign), and SpliceAI predicts no splice alteration (max delta = 0.00). These orthogonal computational lines of evidence support a benign interpretation.3 This variant has been reported in ClinVar (Variation ID 877269) as Uncertain significance by 8 clinical submitters with no expert panel review. No pathogenic or benign assertions have been made by a reputable source. All 16 associated PMIDs are policy or guideline documents unrelated to this specific variant.4 No functional studies (PS3/BS3), case-control data (PS4), de novo observations (PS2/PM6), segregation data (PP1/BS4), or same-residue pathogenic comparators (PS5/PM5) were identified for this variant in the published literature or databases. Applying generic ACMG/AMP 2015 combination rules: PM1_Supporting + PM2_Supporting versus BP4_Supporting results in two opposing supporting-level criteria. Since no criterion reaches moderate or higher strength on either side, and the evidence is balanced with low-confidence signals in both directions, the final classification defaults to Variant of Uncertain Significance (VUS).5
HTRA1
Final classification
VUS
HTRA1 c.1333G>A · p.Ala445Thr
HTRA1
NM_002775.4:c.1333G>A (p.Ala445Thr) is a missense variant in HTRA1 located within the peptidase S1 serine protease domain (residues 157–473), a well-established functional domain critical for HTRA1 protease activity. Loss-of-function missense variants in this domain are a known cause of cerebral small vessel disease including CARASIL and CADASIL2.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 supporting, PM2 supporting, BP4 supporting benign; combination = 2 supporting + 1 supporting benign, which maps to VUS because the evidence is conflicting.
Classification rationale
PM1PM2
BP4
VUS
HTRA1 c.1333G>A
PM1 + PM2 + BP4
→
VUS
1
pvs1_gene_context
3
revelbayesdelspliceai ↗
Gene diagram
· NM_002775.4 · variants mapped to exon structure
HTRA1
NM_002775.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 20 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
supporting
Pathogenic
The variant p.Ala445Thr is located within the peptidase S1 (trypsin-like serine protease) domain (residues 157–473), a well-established critical functional domain. Loss of HTRA1 protease activity through missense variants in this domain is the established mechanism for cerebral small vessel disease (CARASIL/CADASIL2). Published literature confirms that missense variants clustering in the protease domain are a common cause of monogenic CSVD.
UniProt (P83110) annotates the peptidase S1 domain spanning residues 157–473p.Ala445Thr falls within this domain.PMID:28126975: HTRA1 loss of protease activity via mutations in the protease domain causes CARASIL.
✓
PM2
supporting
Pathogenic
This variant is present at very low frequency in population databases. gnomAD v2.1 reports an allele frequency of 0.01237% (35/282,868 alleles, 0 homozygotes) and gnomAD v4.1 reports 0.01041% (168/1,613,814 alleles, 0 homozygotes). Both are well below the 0.1% threshold for PM2. The variant is absent from gnomAD-Canada v1.0. The highest subpopulation frequency is 0.028% (Admixed American, v2.1) and 0.083% (Middle Eastern, v4.1), both below 0.1%.
gnomAD v2.1: AF=0.01237% (35/282868)0 homozygotes
✓
BP4
supporting
Benign
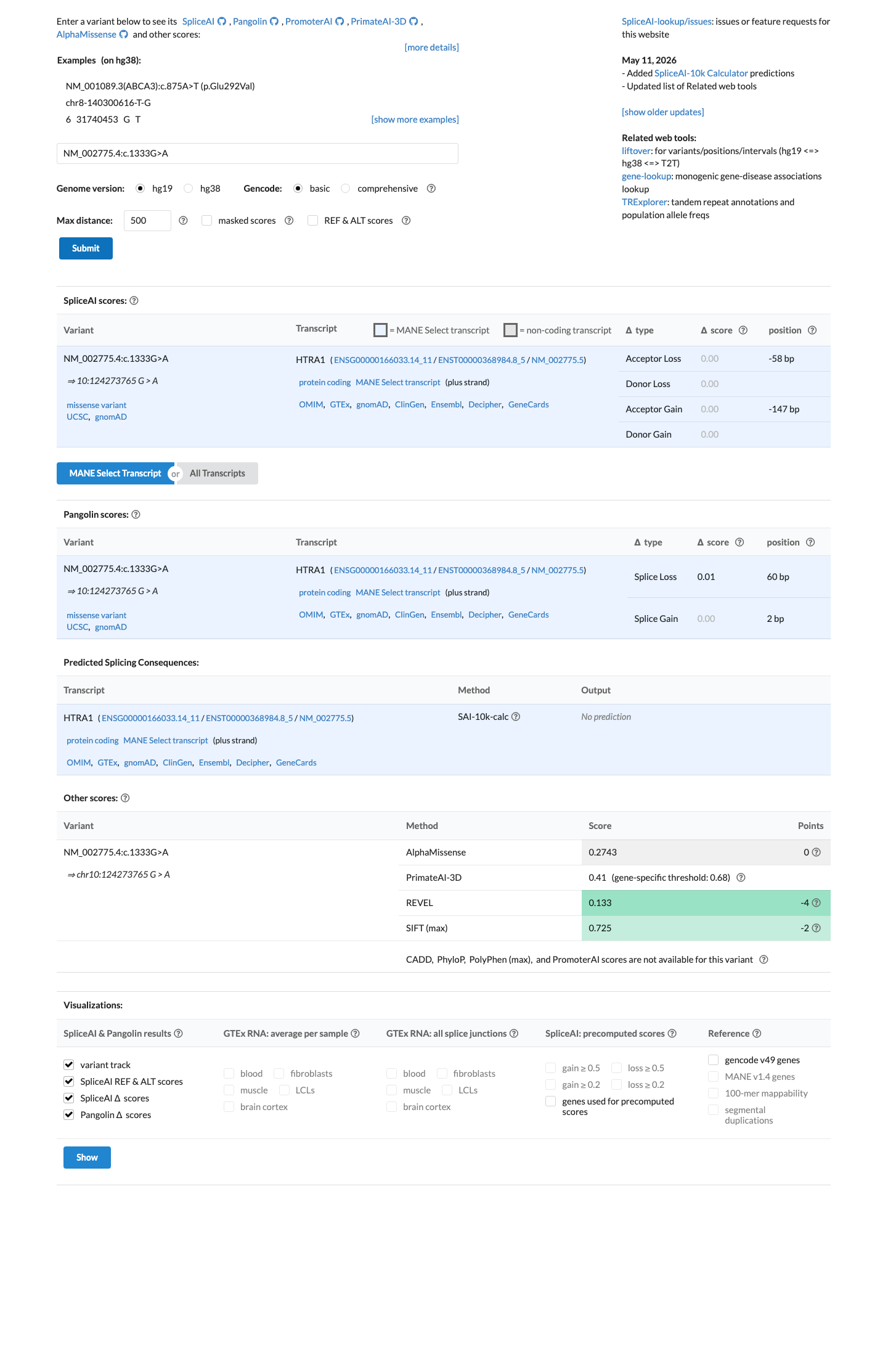
Multiple lines of computational evidence suggest no impact on the gene product. REVEL score is 0.133 (well below pathogenic threshold of 0.5). BayesDel score is -0.219368 (negative, consistent with a benign prediction). SpliceAI predicts no splice alteration (max delta = 0.00). These three independent in silico predictors converge on a neutral/benign interpretation.
REVEL: 0.133 (threshold for pathogenic typically ≥0.5this score is low).BayesDel: -0.219368 (negative score supports a benign interpretation).
Assessed · not applied
Pathogenic
PS1
PS1 requires the same amino acid change (p.Ala445Thr) to have been reported as pathogenic via a different nucleotide change (e.g., c.1333G>C).
PS2
No confirmed de novo occurrence of NM_002775.4:c.1333G>A with established maternity and paternity was identified in the literature or databases.
PS3
No well-established in vitro or in vivo functional studies specifically assessing the damaging effect of p.Ala445Thr on HTRA1 protease activity were identified.
PS4
No case-control study or statistical enrichment analysis has been performed for this variant in HTRA1-associated disease.
PM5
PM5 requires a different pathogenic missense change at the same amino acid residue.
PM6
No assumed de novo occurrence (without confirmation of parentage) has been reported for this variant.
PP1
No co-segregation data for this variant in multiple affected family members were identified.
PP2
PP2 requires a missense variant in a gene with a low rate of benign missense variation where missense is a common disease mechanism.
PP3
Multiple in silico predictors do not support a deleterious effect.
PP4
No detailed patient phenotype or family history data specific to this variant were available.
PP5
No reputable source has classified this variant as pathogenic.
Benign
BA1
BA1 requires an allele frequency >1% in any population database.
BS1
BS1 requires an allele frequency >0.3% in population databases.
BS2
No observation of this variant in a healthy adult individual has been reported for a disorder assumed to be fully penetrant.
BS3
No well-established functional studies demonstrating a neutral effect of p.Ala445Thr on HTRA1 protease activity were identified.
BS4
No evidence of non-segregation with disease in affected members of a family was identified.
BP1
BP1 applies to a missense variant in a gene for which primarily truncating variants are known to cause disease.
BP2
No observation of this variant in trans with a dominant pathogenic variant or in cis with a recessive pathogenic HTRA1 variant was identified.
BP5
No case was identified in which this variant co-occurs with an alternate molecular basis for the observed disease phenotype.
BP6
No reputable source has classified this variant as benign.
N/A · 2
PVS1 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
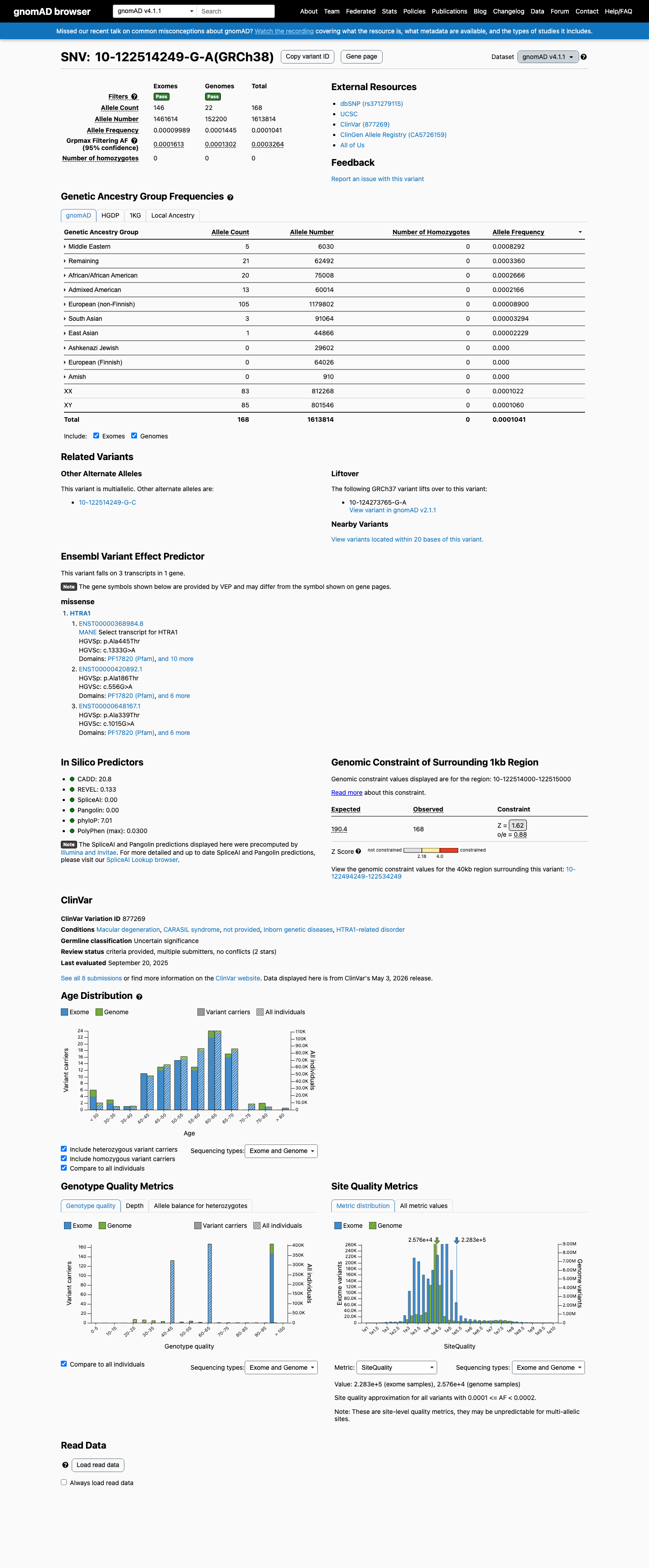
This variant is present in gnomAD v4.1 (AF= 0.000104101; MAF= 0.01041%, 168/1613814 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000829187; MAF= 0.08292%, 5/6030 alleles, homozygotes = 0); grpmax FAF= 0.00032642.
v2.1
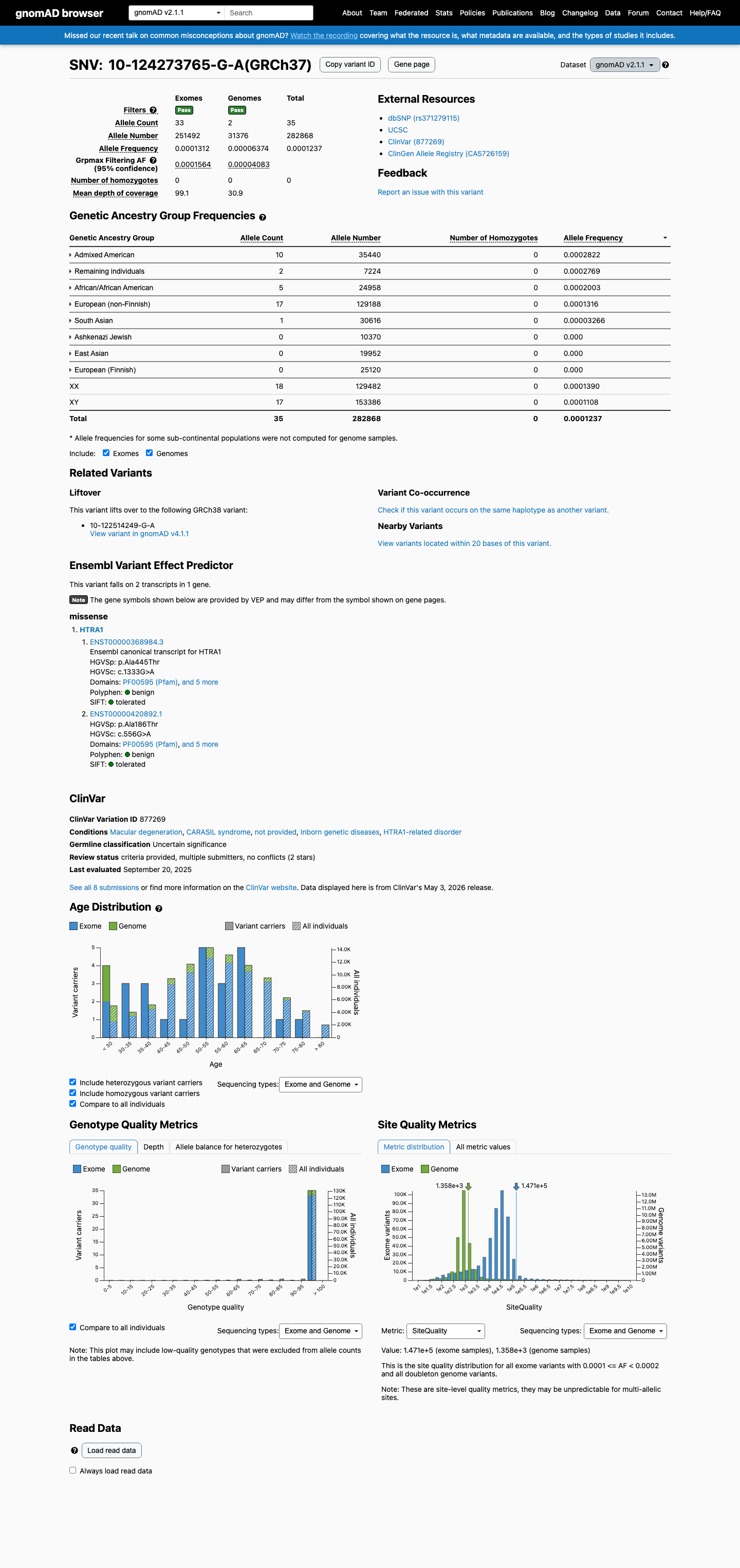
This variant is present in gnomAD v2.1 (AF= 0.000123733; MAF= 0.01237%, 35/282868 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000282167; MAF= 0.02822%, 10/35440 alleles, homozygotes = 0); grpmax FAF= 0.00015644.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.01%
· 168 / 1,613,814
0 hom · FAF 0.033%
0 hom · FAF 0.033%
Middle Eastern 5 / 6,030 |
0.083% |
Remaining individuals 21 / 62,492 |
0.034% |
African/African American 20 / 75,008 |
0.027% |
Admixed American 13 / 60,014 |
0.022% |
European (non-Finnish) 105 / 1,179,802 |
0.0089% |
South Asian 3 / 91,064 |
0.0033% |
East Asian 1 / 44,866 |
0.0022% |
+ 3 not observed (European (Finnish), Amish, Ashkenazi Jewish)
gnomAD v2.1
0.012%
· 35 / 282,868
0 hom · FAF 0.016%
0 hom · FAF 0.016%
Admixed American 10 / 35,440 |
0.028% |
Remaining individuals 2 / 7,224 |
0.028% |
African/African American 5 / 24,958 |
0.02% |
European (non-Finnish) 17 / 129,188 |
0.013% |
South Asian 1 / 30,616 |
0.0033% |
+ 3 not observed (Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (7 clinical laboratories). (ClinVarID = 877269)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.133. BayesDel score = -0.219368.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV64564366, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 14 further PMIDs triaged but not cited — see Sources & References.
PMID 28126975
Found
HTRA1 loss of protease activity via mutations in the protease domain causes CARASIL.
Applied to
→PM1 supports · met
PMID 40607620
Found
Heterozygous HTRA1 missense variants cause autosomal dominant CSVD (CADASIL2) all 15 variants were functionally validated in the protease domain.
Applied to
→PM1 supports · met
Sources & reference links
Triaged references · 14 PMIDs not cited in assessment
23652378 ↗
A framework to start the debate on neonatal screening policies in the EU: an Expert Opinion Document.
CLINVAR
25626707 ↗
Whole-genome sequencing in newborn screening? A statement on the continued importance of targeted approaches in newborn screening programmes.
CLINVAR
25730230 ↗
Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
23169492 ↗
The perspective from EASAC and FEAM on direct-to-consumer genetic testing for health-related purposes.
CLINVAR
22947299 ↗
Specific guidelines for assessing and improving the methodological quality of economic evaluations of newborn screening.
CLINVAR
23037933 ↗
Including the initial newborn screening bloodspot collection device serial number on birth certificates: basis and recommendations from the Secretary of Health and Human Services' Advisory Committee on Heritable Disorders in Newborns and Children.
CLINVAR
23881473 ↗
Newborn screening: education, consent, and the residual blood spot. The position of the national society of genetic counselors.
CLINVAR
24022298 ↗
Offering prenatal diagnostic tests: European guidelines for clinical practice [corrected].
CLINVAR
24394680 ↗
Parental permission for pilot newborn screening research: guidelines from the NBSTRN.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR