NM_000157.4:c.1495G>C (p.Val499Leu) is a missense variant in GBA1, which encodes the lysosomal enzyme glucocerebrosidase. Biallelic pathogenic variants in GBA1 cause Gaucher disease (autosomal recessive); heterozygous variants are associated with increased risk for Parkinson disease. This variant is present at extremely low frequency in population databases: gnomAD v2.1 allele frequency = 0.0052% (13/251,170 alleles, 0 homozygotes) and gnomAD v4.1 allele frequency = 0.0042% (67/1,613,020 alleles, 0 homozygotes), meeting PM2_Supporting.1 In silico analysis supports a deleterious effect: the REVEL meta-predictor score is 0.733, exceeding the commonly used threshold of 0.5. SpliceAI predicts no splicing impact (max delta = 0.00). The BayesDel score (0.348) is below threshold, resulting in partially conflicting computational evidence; PP3 is applied at the supporting level.2 This variant has been reported in ClinVar (Variation ID 634558) with conflicting classifications: Uncertain significance (2 clinical laboratories), Likely pathogenic (2 clinical laboratories), and Likely benign (1 clinical laboratory). No ClinGen expert panel classification is available.3 Functional studies referenced in the literature (PMID:10714667, PMID:12924289) suggest reduced glucocerebrosidase activity for the mature protein equivalent V460L, but full-text verification of these studies was not available at the time of assessment. PS3 is not assessed pending direct review of primary data. The ClinGen Parkinson's Disease Expert Panel specification for GBA1 (v1.0.0) is an unstructured ruleset without specific criterion thresholds. The assessment therefore applies generic ACMG/AMP 2015 criteria (PMID:25741868).4 Overall, the evidence applied includes PM2_Supporting and PP3_Supporting. No benign criteria were met. All remaining criteria (PVS1, PS1-PS5, PM1, PM5-PM6, PP1-PP2, PP4-PP5, BA1, BS1-BS4, BP1-BP2, BP4-BP7) were either not met, not assessed, or not applicable.
GBA1
Final classification
VUS
GBA1 c.1495G>C · p.Val499Leu
GBA1
NM_000157.4:c.1495G>C (p.Val499Leu) is a missense variant in GBA1, which encodes the lysosomal enzyme glucocerebrosidase. Biallelic pathogenic variants in GBA1 cause Gaucher disease (autosomal recessive); heterozygous variants are associated with increased risk for Parkinson disease.
ClinGen Parkinson's Disease Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for GBA1 Version 1.0.0 v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, PP3 supporting; combination = 2 supporting, which maps to VUS.
Classification rationale
PM2PP3
VUS
GBA1 c.1495G>C
PM2 + PP3
→
VUS
Gene diagram
· NM_000157.4 · variants mapped to exon structure
GBA1
NM_000157.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in population databases: gnomAD v2.1 allele frequency = 5.18e-05 (13/251,170 alleles, 0 homozygotes, grpmax FAF = 5.60e-05) and gnomAD v4.1 allele frequency = 4.15e-05 (67/1,613,020 alleles, 0 homozygotes, grpmax FAF = 3.75e-05). Both are well below the 0.1% PM2 threshold. The variant is absent from gnomAD-Canada v1.0.
gnomAD v2.1: AF = 0.0052%13/251170 alleles
✓
PP3
supporting
Pathogenic
Multiple lines of computational evidence support a deleterious effect. The REVEL meta-predictor score is 0.733, which exceeds the commonly used threshold of 0.5 for predicting pathogenicity. SpliceAI predicts no splicing impact (max delta = 0.00), which is neutral for this criterion. The BayesDel score is 0.348 (below the 0.5 threshold), but the REVEL score provides sufficient in silico support at the supporting level.
REVEL score = 0.733 (>0.5 threshold)BayesDel score = 0.348 (<0.5 threshold)SpliceAI max delta = 0.00 (no splicing impact)
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change at the same codon resulting in the same amino acid change (p.Val499Leu) with established pathogenicity.
PS2
PS2 requires a de novo occurrence with confirmed maternity and paternity.
PS3
The exploratory evidence pipeline identified functional studies (PMID:10714667 Koprivica et al.
PS4
PS4 requires statistically significant enrichment of the variant in affected individuals compared to controls.
PM1
PM1 requires location in a mutational hotspot or critical functional domain without benign variation.
PM6
PM6 requires a de novo occurrence with confirmed maternity and paternity.
PP1
PP1 requires cosegregation with disease in multiple affected family members.
PP2
PP2 requires a low rate of benign missense variation in the gene and that missense variants are a common mechanism of disease.
PP4
PP4 requires that the patient's phenotype or family history is highly specific for the disease.
PP5
PP5 requires that a reputable source has classified the variant as pathogenic.
Benign
BA1
BA1 requires allele frequency >1% in population databases.
BS1
BS1 requires allele frequency >0.3% in population databases.
BS2
BS2 requires observation in a healthy adult in the homozygous state for a recessive disorder.
BS3
BS3 requires well-established in vitro or in vivo functional studies showing no damaging effect.
BS4
BS4 requires lack of segregation in affected family members.
BP1
BP1 applies to missense variants in genes where primarily truncating variants cause disease.
BP2
BP2 requires observation in trans with a pathogenic variant for a dominant disorder, or in cis with a pathogenic variant in a recessive disorder in an unaffected individual.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene product.
BP5
BP5 requires that the variant is found in a case with an alternate molecular basis for disease.
BP6
BP6 requires that a reputable source classifies the variant as benign.
N/A · 4
PVS1 · PM5 · BP3 · BP7
Research & evidence
Population frequency
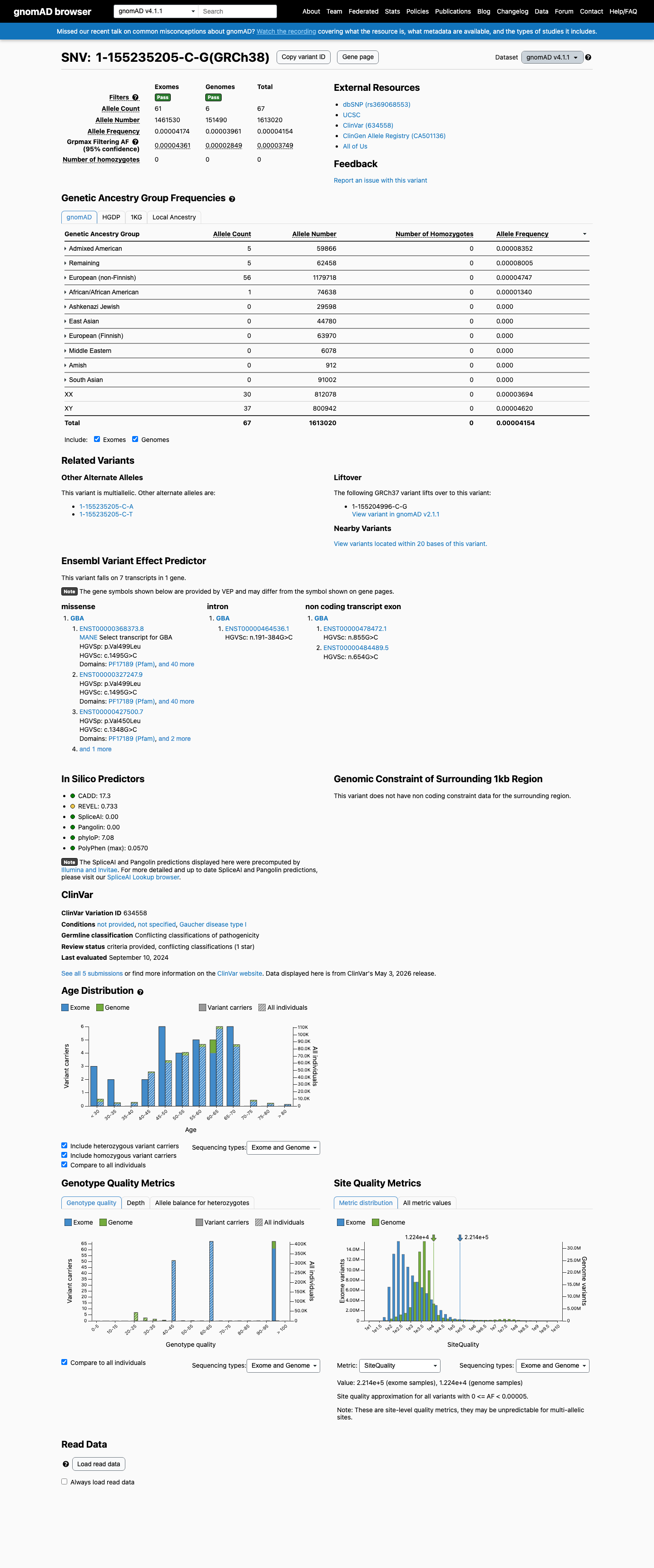
gnomAD v4.1
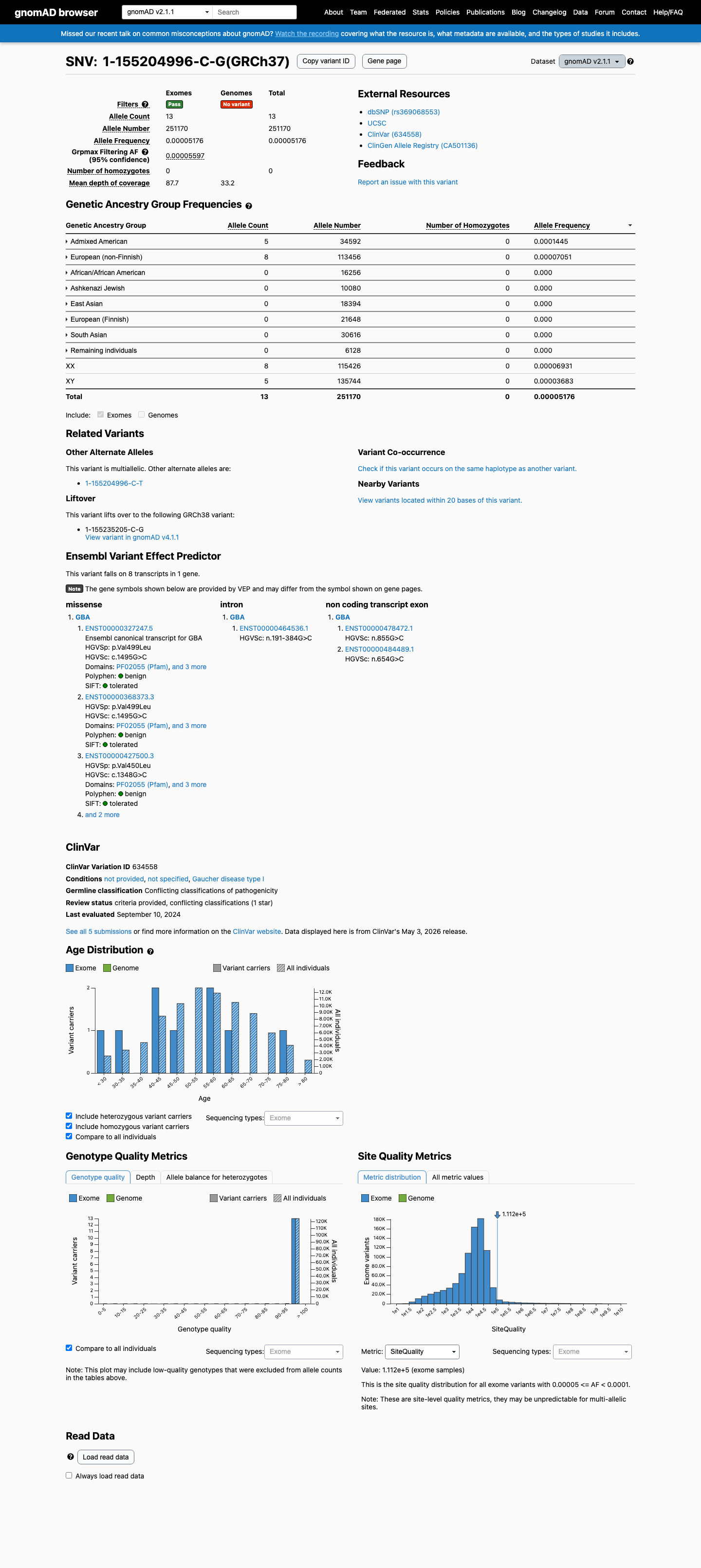
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 4.1537e-05; MAF= 0.00415%, 67/1613020 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 8.35199e-05; MAF= 0.00835%, 5/59866 alleles, homozygotes = 0); grpmax FAF= 3.749e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 5.17578e-05; MAF= 0.00518%, 13/251170 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000144542; MAF= 0.01445%, 5/34592 alleles, homozygotes = 0); grpmax FAF= 5.597e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0042%
· 67 / 1,613,020
0 hom · FAF 0.0037%
0 hom · FAF 0.0037%
Admixed American 5 / 59,866 |
0.0084% |
Remaining individuals 5 / 62,458 |
0.008% |
European (non-Finnish) 56 / 1,179,718 |
0.0047% |
African/African American 1 / 74,638 |
0.0013% |
+ 6 not observed (European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0052%
· 13 / 251,170
0 hom · FAF 0.0056%
0 hom · FAF 0.0056%
Admixed American 5 / 34,592 |
0.014% |
European (non-Finnish) 8 / 113,456 |
0.0071% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
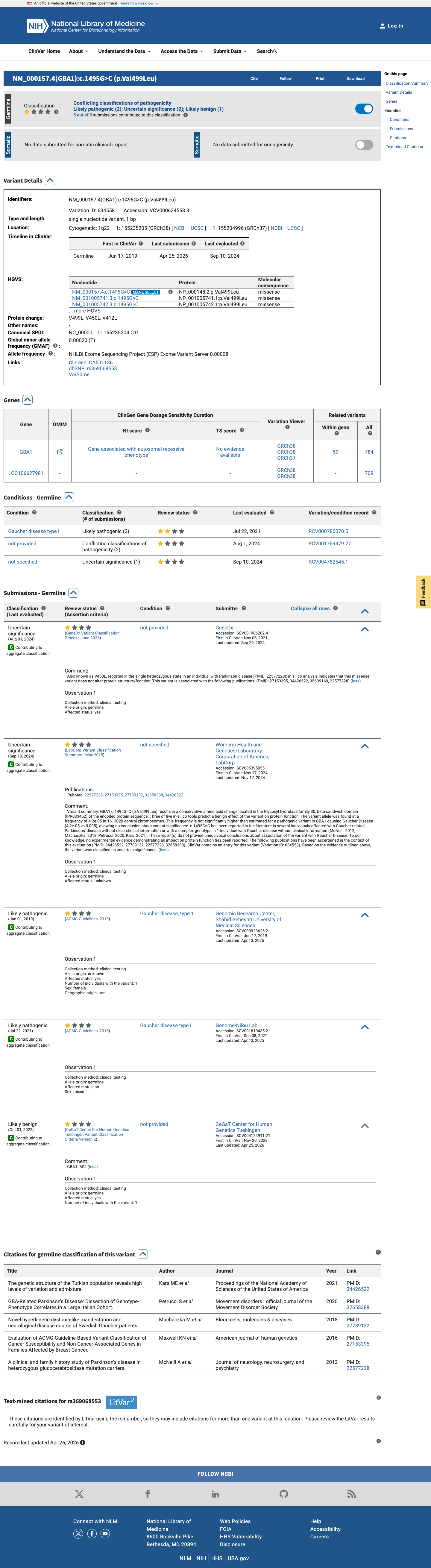
ClinVar
This variant has been reported in ClinVar as Uncertain significance (2 clinical laboratories) and as Likely pathogenic (2 clinical laboratories) and as Likely benign (1 clinical laboratory). (ClinVarID = 634558)
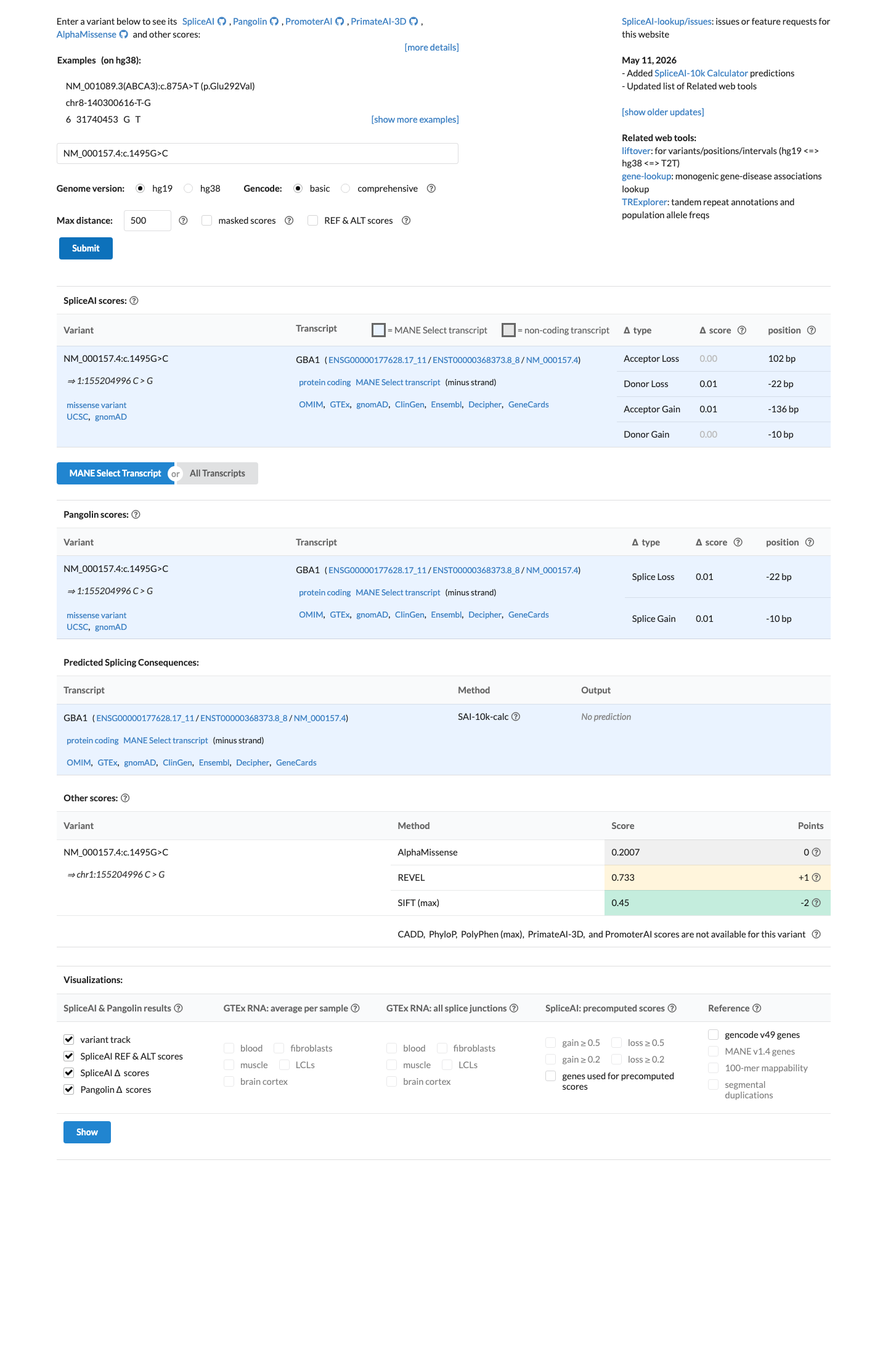
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.733. BayesDel score = 0.348057.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
27153395 ↗
Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer.
CLINVAR
32658388 ↗
GBA-Related Parkinson's Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort.
CLINVAR
34426522 ↗
The genetic structure of the Turkish population reveals high levels of variation and admixture.
CLINVAR
19888064 ↗
ACOG Committee Opinion No. 442: Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent.
CLINVAR
22577228 ↗
A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers.
CLINVAR