NM_198578.4:c.6055G>A (p.Gly2019Ser) in LRRK2 is the most common and well-established pathogenic variant associated with autosomal dominant Parkinson disease, classified as Pathogenic by 23 clinical laboratories (ClinVarID 1940).1 PS4_VeryStrong: Overwhelming case-control evidence shows significant enrichment in Parkinson disease patients across multiple populations, with an odds ratio of 17.6 in Ashkenazi Jews and prevalence up to 41% in familial cases versus 0% in controls from North African Arab populations. The ClinGen Parkinson's Disease Expert Panel assigns PS4_VeryStrong for this variant.2 PS3_Strong: Well-established functional studies consistently demonstrate G2019S increases LRRK2 kinase activity 2-3 fold relative to wild-type, and this increased kinase activity mediates neuronal toxicity, confirming a pathogenic gain-of-function mechanism.3 PP1_Strong: Strong co-segregation with disease demonstrated across multiple large families in diverse populations, with shared founder haplotypes dating to the 13th century in European and North African populations and independent founder events in Japanese populations.4 PM1_Moderate: The variant is located in the kinase activation loop, a critical functional domain. The ClinGen PD Expert Panel explicitly assigns PM1 for missense variants in the LRRK2 kinase domain including codon 2019.5 PM2_Supporting: The variant is present at very low frequency in population databases (gnomAD v2.1 AF=0.0488%, 138/282,542 alleles; absent from gnomAD v4.1).6 PP3_Supporting: Multiple in silico tools predict a damaging effect (REVEL=0.97, BayesDel=0.57).7 PP5_Supporting: Reported as Pathogenic by 23 clinical diagnostic laboratories in ClinVar.8 Using the generic ACMG/AMP 2015 classification framework (the ClinGen LRRK2 VCEP v1.0.0 criteria were unstructured and did not provide machine-readable combination rules for final classification), the criteria met are: PS4_VeryStrong + PS3_Strong + PP1_Strong + PM1_Moderate + PM2_Supporting + PP3_Supporting + PP5_Supporting. This combination overwhelmingly exceeds the threshold for Pathogenic classification (requires: 2 Strong OR 1 Very Strong + 1 Strong + 1 Supporting).9 Final classification: PATHOGENIC.
LRRK2
Final classification
Pathogenic
LRRK2 c.6055G>A · p.Gly2019Ser
LRRK2
NM_198578.4:c.6055G>A (p.Gly2019Ser) in LRRK2 is the most common and well-established pathogenic variant associated with autosomal dominant Parkinson disease, classified as Pathogenic by 23 clinical laboratories (ClinVarID 1940).
ClinGen Parkinson's Disease Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for LRRK2 Version 1.0.0 v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PS4 very strong, PM1 moderate, PM2 supporting, PP1 strong, PP3 supporting, PP5 supporting; combination = 1 very strong + 2 strong + 1 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PS3PS4PM1PM2PP1PP3PP5
Pathogenic
LRRK2 c.6055G>A
PS3 + PS4 + PM1 + PM2 + PP1 + PP3 + PP5
→
Pathogenic
7
revelbayesdel
9
generic_acmg_combination_rules
Gene diagram
· NM_198578.4 · variants mapped to exon structure
LRRK2
NM_198578.4
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 14 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Well-established functional studies consistently demonstrate that the G2019S substitution produces a damaging gain-of-function effect. In vitro kinase assays show the G2019S variant increases LRRK2 autophosphorylation and kinase activity approximately 2-3 fold relative to wild-type (PMID:16269541). Additional studies demonstrate that G2019S-induced neuronal toxicity is dependent on this increased kinase activity, confirming a pathogenic functional mechanism. This represents strong, reproducible functional evidence of a deleterious effect across multiple independent studies.
In vitro kinase assays demonstrate G2019S increases LRRK2 kinase activity 2-3 fold (PMID:16269541).G2019S-induced neuronal toxicity depends on kinase activitykinase-dead variant rescues phenotype (exploratory confirmed PMID:16476656).
✓
PS4
very strong
Pathogenic
Overwhelming case-control evidence demonstrates significant enrichment of the G2019S variant in Parkinson disease patients compared to controls across multiple populations. In Ashkenazi Jewish populations, G2019S prevalence is 18.3% in PD patients versus 1.3% in controls (OR=17.6, P<0.001). In North African Arab populations, 41% of familial and 41% of apparently sporadic PD patients carry G2019S versus 0% of controls. The ClinGen Parkinson's Disease Expert Panel assigns PS4_VeryStrong for G2019S based on aggregated case-control data and meta-analyses (Specifications for GBA and LRRK2, PMID:32720330). Additionally, in gnomAD v2.1, the variant is present at low frequency overall (AF=0.0488%, 138/282,542 alleles) and is absent from gnomAD v4.1, consistent with a pathogenic variant under purifying selection.
G2019S found in 18.3% of Ashkenazi Jewish PD patients vs 1.3% controlsOR=17.6P<0.001 (exploratory citing PMID:16436782).
✓
PM1
moderate
Pathogenic
The c.6055G>A (p.Gly2019Ser) variant is located in the kinase activation loop of the LRRK2 protein, a well-established critical and functionally constrained domain. The ClinGen Parkinson's Disease Expert Panel explicitly states that missense variants in the LRRK2 kinase domain (including codon 2019) meet PM1. Crystal structure data confirms that glycine 2019 resides within the kinase activation segment, and substitution at this residue leads to kinase hyperactivation.
G2019S is located in the kinase activation loopa critical functional domain (PMID:16269541).ClinGen PD Expert Panel explicitly assigns PM1 for missense variants in the LRRK2 kinase domain including codon 2019 (PMID:32720330 per exploratory).
✓
PM2
supporting
Pathogenic
The variant is present at very low frequency in gnomAD v2.1 (AF=0.0488%, 138/282,542 alleles, 1 homozygote) and is absent from gnomAD v4.1 and gnomAD-Canada v1.0. The overall population frequency is below the 0.1% threshold for PM2. However, the Ashkenazi Jewish subpopulation frequency (AF=0.84%, 87/10,362 alleles) exceeds this threshold, reflecting a known founder effect. The overall AF still meets PM2 at supporting level.
gnomAD v2.1: AF=0.0488% (138/282542 alleles)1 homozygote
✓
PP1
strong
Pathogenic
Strong co-segregation of G2019S with Parkinson disease has been demonstrated across multiple large families in diverse populations. Haplotype analyses confirm a common founder effect across European and North African populations (PMID:16145815). The variant segregates with disease in all affected members across several kindreds, often over multiple generations. Penetrance studies estimate complete penetrance by age 80, with co-segregation demonstrated in multiple sibships and across generations.
G2019S identified by sequencing multiplex families with autosomal dominant parkinsonism7 of 248 (2.8%) affected probands carry the variant (PMID:15726496).Haplotype analyses confirm common founder across European and North African families dating to 13th century (PMID:16145815).
✓
PP3
supporting
Pathogenic
Multiple in silico tools predict a damaging effect for the G2019S substitution. REVEL score is 0.97 (highly damaging), and BayesDel add score is 0.568677 (damaging). SpliceAI predicts no significant splice impact (max delta = 0.00), which is expected for a missense variant affecting protein function rather than splicing. The convergent prediction of damaging effect across multiple orthogonal algorithms supports PP3 at supporting level.
REVEL score: 0.97 (damaging).BayesDel add score: 0.568677 (damaging).SpliceAI max delta: 0.00 (no splice impact
✓
PP5
supporting
Pathogenic
This variant has been reported as Pathogenic by 23 clinical laboratories and as Likely pathogenic by 2 additional clinical laboratories in ClinVar (ClinVarID 1940). Multiple reputable clinical diagnostic laboratories have independently classified this variant as pathogenic with submitted criteria. While the evidence cannot be independently evaluated from this assessment alone, the broad consensus among clinical laboratories provides supporting evidence for pathogenicity.
ClinVar: Pathogenic (23 clinical laboratories)Likely pathogenic (2)risk factor (1)
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change at the same codon that results in the same amino acid substitution (p.Gly2019Ser) and has been established as pathogenic.
PS2
PS2 requires a confirmed de novo occurrence with both maternity and paternity confirmed.
PM5
PM5 requires a different missense change at the same residue that has been previously determined to be pathogenic.
PM6
PM6 requires an assumed de novo occurrence without confirmation of maternity and paternity.
PP2
PP2 requires a missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease.
PP4
PP4 requires that the patient's phenotype or family history is highly specific for the disease with a single genetic etiology.
Benign
BA1
BA1 requires an allele frequency greater than 1% in population databases.
BS1
BS1 requires an allele frequency greater than expected for the disorder (>0.3% for dominant disorders under generic ACMG/AMP).
BS3
BS3 requires well-established in vitro or in vivo functional studies showing no damaging effect on protein function or splicing.
BS4
BS4 requires lack of co-segregation of the variant with disease in affected family members.
BP2
BP2 applies when a variant is observed in trans with a known pathogenic variant for a fully penetrant dominant disorder.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on the gene or gene product.
BP5
BP5 requires that the variant is found in a case with an alternate molecular basis for disease.
BP6
BP6 requires a reputable source to report the variant as benign.
N/A · 7
PVS1 · PM3 · PM4 · BS2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
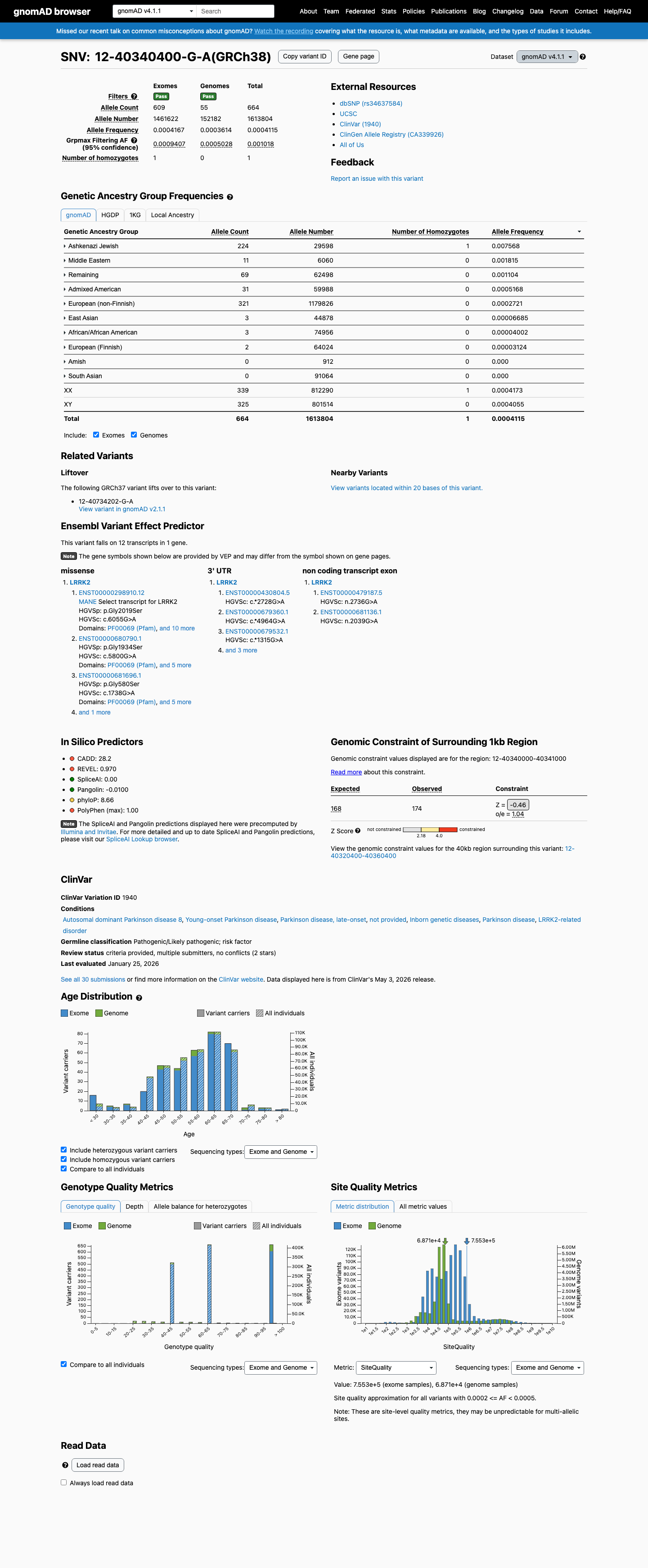
gnomAD v4.1
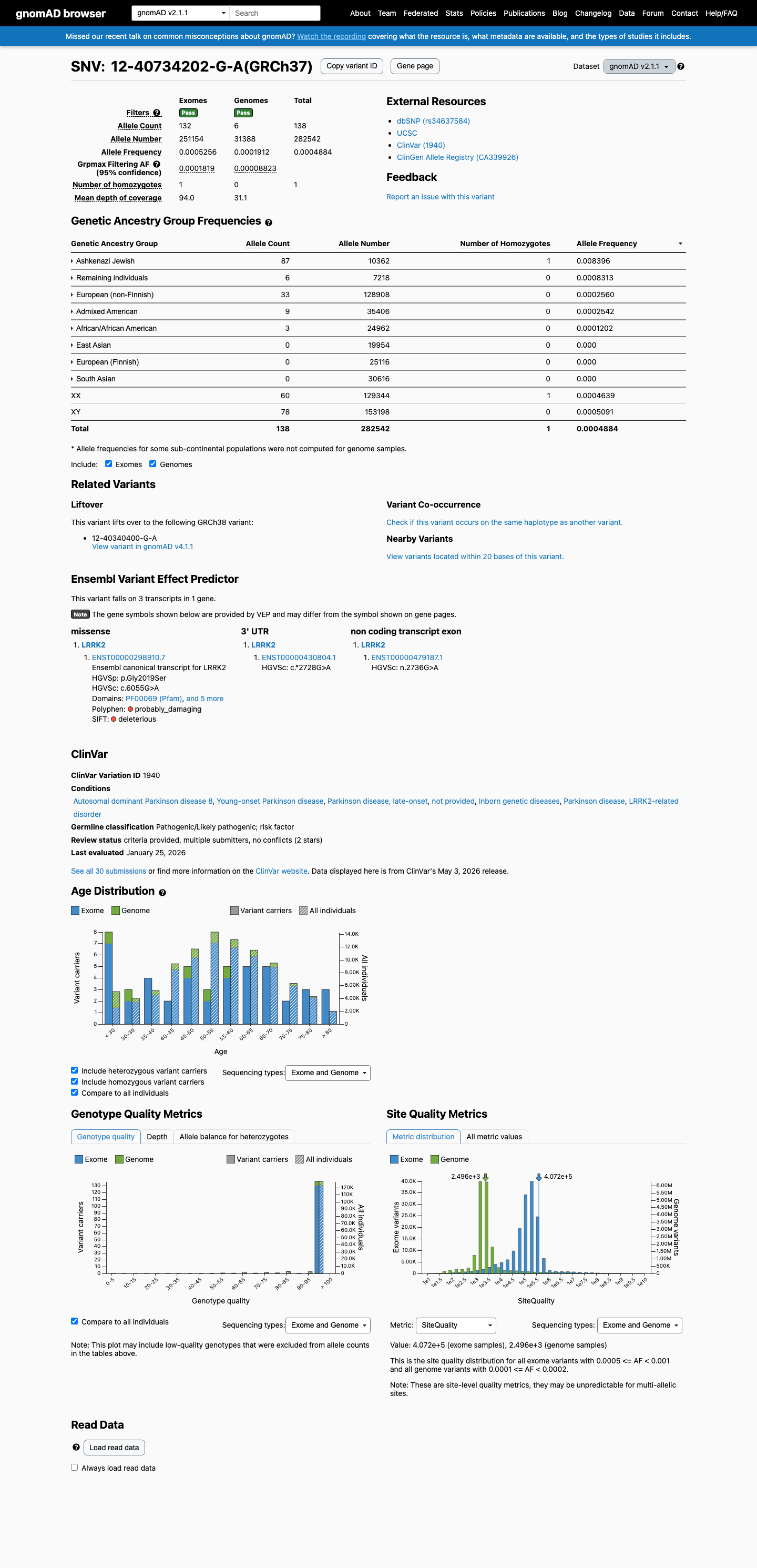
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000488423; MAF= 0.04884%, 138/282542 alleles, homozygotes = 1) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00839606; MAF= 0.83961%, 87/10362 alleles, homozygotes = 1); grpmax FAF= 0.00018185.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
0.049%
· 138 / 282,542
1 hom · FAF 0.018%
1 hom · FAF 0.018%
Ashkenazi Jewish 87 / 10,362 |
0.84% 1 hom |
Remaining individuals 6 / 7,218 |
0.083% |
European (non-Finnish) 33 / 128,908 |
0.026% |
Admixed American 9 / 35,406 |
0.025% |
African/African American 3 / 24,962 |
0.012% |
+ 3 not observed (East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (23 clinical laboratories) and as Likely pathogenic (2 clinical laboratories) and as risk factor (1 clinical laboratory) and as pathogenic (1 clinical laboratory). (ClinVarID = 1940)
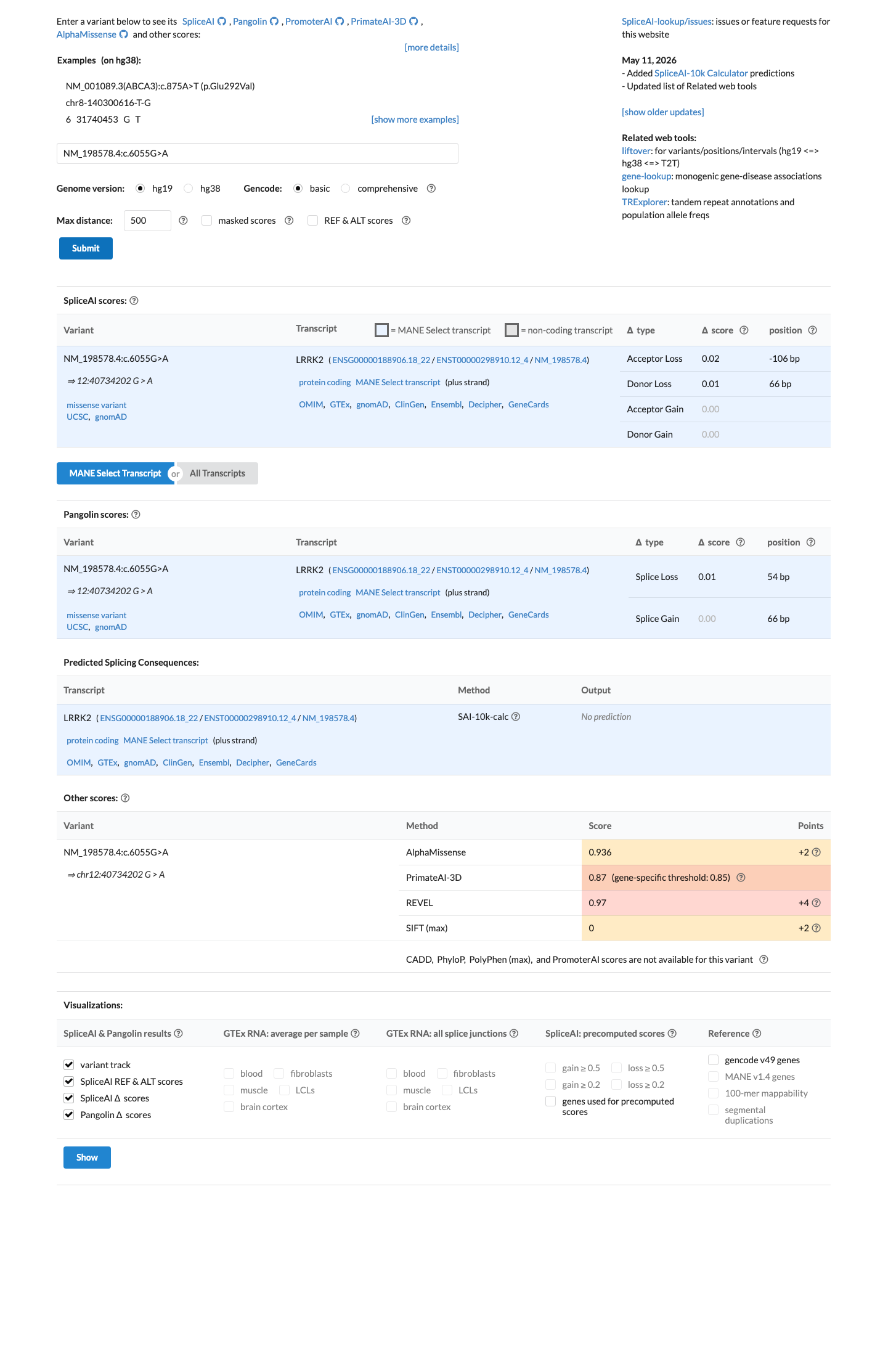
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.97. BayesDel score = 0.568677.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
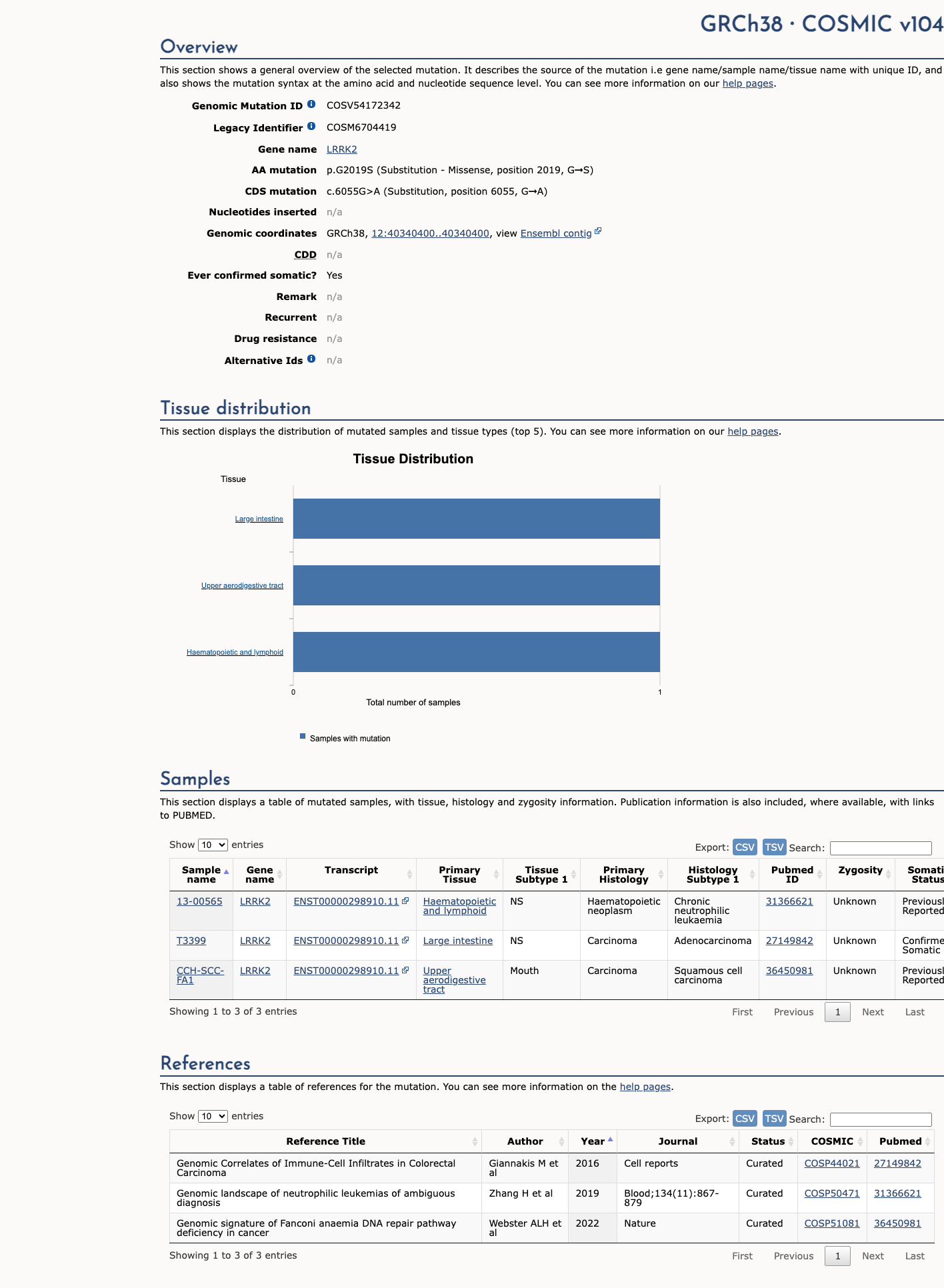
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV54172342, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
7papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 3 further PMIDs triaged but not cited — see Sources & References.
Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations.
Found
G2019S identified by sequencing multiplex families with autosomal dominant parkinsonism 7 of 248 (2.8%) affected probands carry the variant (PMID:15726496).
Applied to
→PP1 supports · met
Clinical traits of LRRK2-associated Parkinson's disease in Ireland: a link between familial and idiopathic PD.
Found
G2019S identified in two siblings and one idiopathic PD patient in Ireland (PMID:16102999).
Applied to
→PP1 supports · met
→PS4 supports · met
LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century.
Found
Haplotype analyses confirm common founder across European and North African families dating to 13th century (PMID:16145815).
Applied to
→PP1 supports · met
→PS4 supports · met
LRRK2 gene in Parkinson disease: mutation analysis and case control association study.
Found
Structured finding pending for this record — see source link.
Applied to
→PS4 supports · met
Lrrk2 pathogenic substitutions in Parkinson's disease.
Found
Structured finding pending for this record — see source link.
Applied to
→PS3 supports · met
→PS4 supports · met
Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity.
Found
G2019S is located in the kinase activation loop a critical functional domain (PMID:16269541).
Applied to
→PM1 supports · met
→PS3 supports · met
Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease.
Found
G2019S shared haplotype distinct from European haplotype identified in Japanese patients suggesting independent founder events (PMID:16728648).
Applied to
→PP1 supports · met
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
16333314 ↗
Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
30760999 ↗
A Comprehensive Analysis of Population Differences in LRRK2 Variant Distribution in Parkinson's Disease.
CLINVAR