NM_000059.4:c.5946del is a frameshift deletion in BRCA2 exon 11 resulting in a premature termination codon (p.Ser1982ArgfsTer22) with expected nonsense-mediated decay, in a gene where loss of function is an established mechanism for hereditary breast and ovarian cancer. Per ENIGMA Specifications Table 4, BRCA2 exon 11 PTC variants are assigned PVS1 at very strong weight.1 ENIGMA Specifications Table 4 assigns PM5_Strong (PTC) to BRCA2 exon 11, indicating additional pathogenic weight for a protein termination codon variant in an exon where other proven pathogenic PTC variants have been previously observed.2 Clinical-history likelihood ratio analysis from Li et al. 2020 (PMID:31853058) yields an LR of 31.79 based on 149 carriers, exceeding the ENIGMA PP4_Strong threshold of ≥18.7:1. The personal and family cancer history profile of individuals carrying this variant is significantly enriched for breast and ovarian cancer compared to non-carriers.3 The variant is present in gnomAD population databases at low frequency in non-founder populations (grpmax FAF=5.39e-05 in v2.1, 2.154e-05 in v4.1), meeting ENIGMA BS1_Supporting (FAF >0.002% and ≤0.01%). The higher Ashkenazi Jewish allele frequency (0.589%) is attributable to a known founder effect and is excluded from BS1/BA1 assessment per ENIGMA non-founder population rules.4 This variant has been reported in ClinVar as Pathogenic by 67 clinical laboratories and by the ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel (ClinVar Variation ID: 9325). The variant has been observed in multiple affected individuals across diverse populations and has been described as a recurrent pathogenic founder mutation.5 Under ENIGMA Table 3 combining rules, the combination of PVS1 (Very Strong) + PM5 (Strong) satisfies the Pathogenic classification threshold (1 Very Strong + ≥1 Strong). PP4 (Strong) provides additional corroborating evidence. BS1_Supporting does not alter the pathogenic classification.6
BRCA2
Final classification
Pathogenic
BRCA2 c.5946del · p.Ser1982ArgfsTer22
BRCA2
NM_000059.4:c.5946del is a frameshift deletion in BRCA2 exon 11 resulting in a premature termination codon (p.Ser1982ArgfsTer22) with expected nonsense-mediated decay, in a gene where loss of function is an established mechanism for hereditary breast and ovarian cancer. Per ENIGMA Specifications Table 4, BRCA2 exon 11 PTC variants are assigned PVS1 at very strong weight.
ENIGMA BRCA1/BRCA2 v1.2.0 Table 3 point system (+16: PVS1+8, PM5+4, PP4+4, PP5+1, BS1-1) meets pathogenic threshold >=10; all_of rule 1 Very Strong + >=1 Strong also independently satisfied.
Classification rationale
PVS1PM5PP4PP5
BS1
Pathogenic
BRCA2 c.5946del
PVS1 + PM5 + PP4 + PP5 + BS1
→
Pathogenic
1
vcep_specifications_table4_v1_2_2024_11_18cspec ↗pvs1_generic_framework ↗
2
vcep_specifications_table4_v1_2_2024_11_18cspec ↗
3
vcep_pmid_31853058_brca2_clinical_history_lrPMID:31853058 ↗
6
cspec ↗
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 9 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_000059.4:c.5946del is a frameshift deletion in BRCA2 exon 11 predicted to cause a premature termination codon (p.Ser1982ArgfsTer22) with expected nonsense-mediated decay. Loss of function is an established disease mechanism for BRCA2 in hereditary breast and ovarian cancer. Per ENIGMA Specifications Table 4, BRCA2 exon 11 PTC variants are assigned PVS1 at very strong weight; the variant occurs upstream of the DNA-binding domain (aa 2481-3186) and is not in a PVS1-downgraded region.
Frameshift deletion (c.5946del) producing p.Ser1982ArgfsTer22 in BRCA2 exon 11ENIGMA Table 4 assigns PVS1 at very strong weight for BRCA2 E11 PTC variantsNMD expected
✓
PM5
strong
Pathogenic
This is a protein termination codon (PTC) variant in BRCA2 exon 11. Per ENIGMA Specifications Table 4, exon 11 is assigned PM5_Strong (PTC), indicating that other proven pathogenic PTC variants have been previously observed in this exon. The variant qualifies for PVS1 and PM5_PTC provides additional weight.
ENIGMA Table 4: BRCA2 E11 assigned PM5_Strong (PTC)E11 is not a PM5_N/A exon (PM5_N/A exons: E12E27
✓
PP4
strong
Pathogenic
Clinical-history likelihood ratio from Li et al. 2020 (PMID:31853058) is LR=31.79 based on 149 probands. This exceeds the ENIGMA PP4_Strong threshold of ≥18.7:1, indicating that the personal and family cancer history profile of carriers is significantly enriched for pathogenicity.
Li et al. 2020 clinical-history LR = 31.79 (149 probands)Exceeds PP4_Strong threshold of LR ≥18.7:1Personal/family cancer history profile consistent with pathogenic BRCA2 variant
✓
PP5
supporting
Pathogenic
Expert panel ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel, ClinGen classified as Pathogenic.
ClinVar expert panel classification
✓
BS1
supporting
Benign
ENIGMA BS1_Supporting applies when FAF > 0.002% and ≤0.01% in gnomAD non-founder populations. The grpmax FAF is 5.39e-05 (0.00539%) in gnomAD v2.1, which falls within this range. The variant is observed in outbred population controls at a low but non-negligible frequency, providing supporting evidence that it may be more common than expected for a highly penetrant pathogenic variant.
gnomAD v2.1 grpmax FAF = 5.39e-05 (0.00539%)meets BS1_Supporting range (>0.002%≤0.01%)
Assessed · not applied
Pathogenic
PS3
ENIGMA Table 9 (curated functional assay results) applies to missense and synonymous variants and contains no entry for this frameshift PTC variant.
PS4
ENIGMA PS4 requires a formal case-control study with p-value ≤0.05, OR ≥4, and lower CI excluding 2.0.
PM2
ENIGMA PM2_Supporting requires absence from gnomAD v2.1 (non-cancer, exome) and gnomAD v3.1 (non-cancer) in outbred populations.
PP1
ENIGMA PP1 requires quantitative co-segregation analysis with LR thresholds (Supporting ≥2.08, Moderate ≥4.3, Strong ≥18.7).
Benign
BA1
ENIGMA BA1 (Stand-Alone benign) requires FAF > 0.1% in gnomAD non-founder populations.
BS2
ENIGMA BS2 is applied in the absence of features of recessive Fanconi anemia (FA) phenotype in individuals carrying the variant.
BS3
ENIGMA BS3 applies when well-established functional studies show no damaging effect on protein function.
BS4
ENIGMA BS4 requires quantitative lack-of-segregation analysis (LR ≤0.48 for Supporting).
BP5
ENIGMA BP5 captures clinical-history LR against pathogenicity.
N/A · 13
PS1 · PS2 · PM1 · PM4 · PM6 · PP2 · PP3 · BP1 · BP2 · BP3 · BP4 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic

gnomAD v4.1
gnomAD v2.1
v4.1
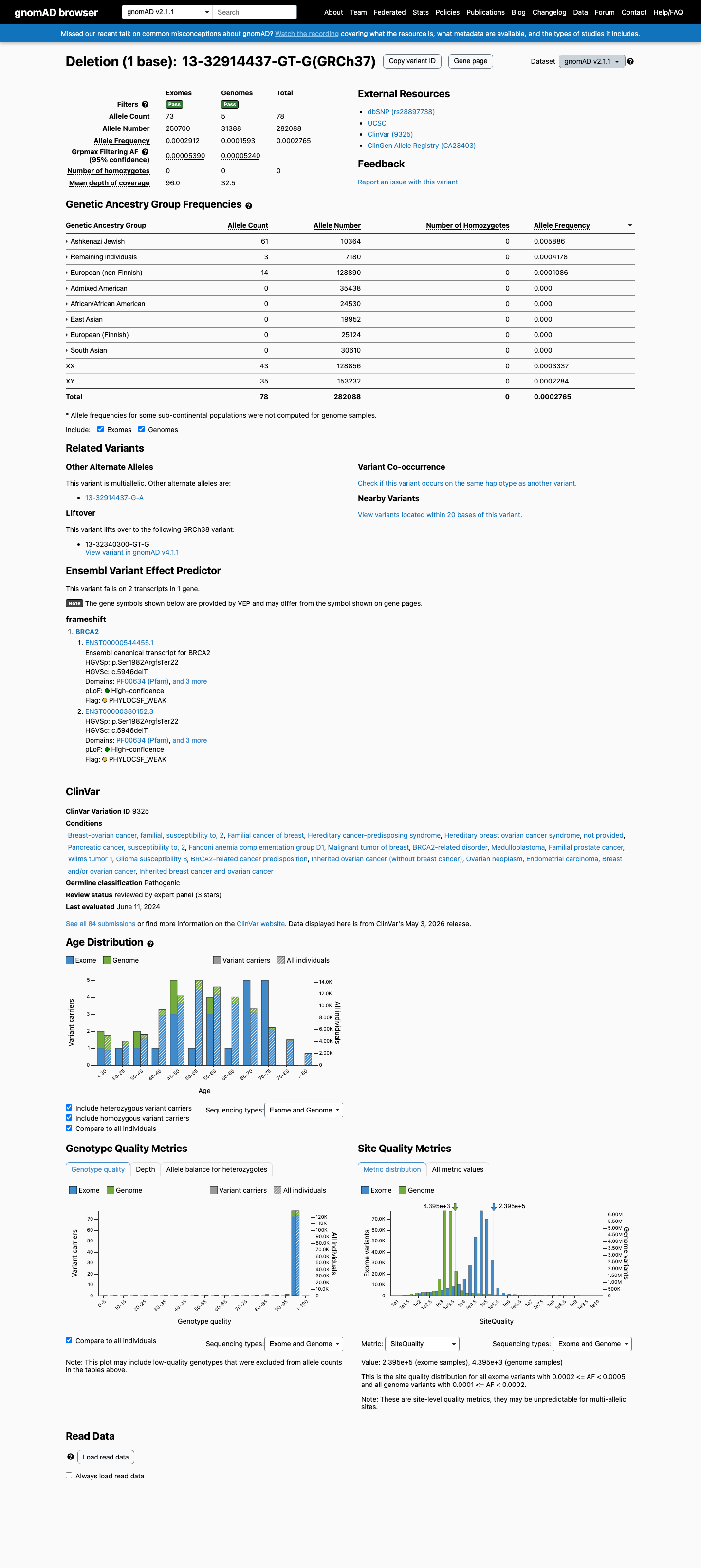
This variant is present in gnomAD v4.1 (AF= 0.000139416; MAF= 0.01394%, 225/1613878 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00533856; MAF= 0.53386%, 158/29596 alleles, homozygotes = 0); grpmax FAF= 2.154e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000276509; MAF= 0.02765%, 78/282088 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00588576; MAF= 0.58858%, 61/10364 alleles, homozygotes = 0); grpmax FAF= 5.39e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.014%
· 225 / 1,613,878
0 hom · FAF 0.0022%
0 hom · FAF 0.0022%
Ashkenazi Jewish 158 / 29,596 |
0.53% |
Remaining individuals 31 / 62,478 |
0.05% |
European (non-Finnish) 35 / 1,179,918 |
0.003% |
African/African American 1 / 74,912 |
0.0013% |
+ 6 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian)
gnomAD v2.1
0.028%
· 78 / 282,088
0 hom · FAF 0.0054%
0 hom · FAF 0.0054%
Ashkenazi Jewish 61 / 10,364 |
0.59% |
Remaining individuals 3 / 7,180 |
0.042% |
European (non-Finnish) 14 / 128,890 |
0.011% |
+ 5 not observed (African/African American, Admixed American, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (67 clinical laboratories) and as pathogenic (1 clinical laboratory) and as Pathogenic by ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel, ClinGen (expert panel). (ClinVarID = 9325)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
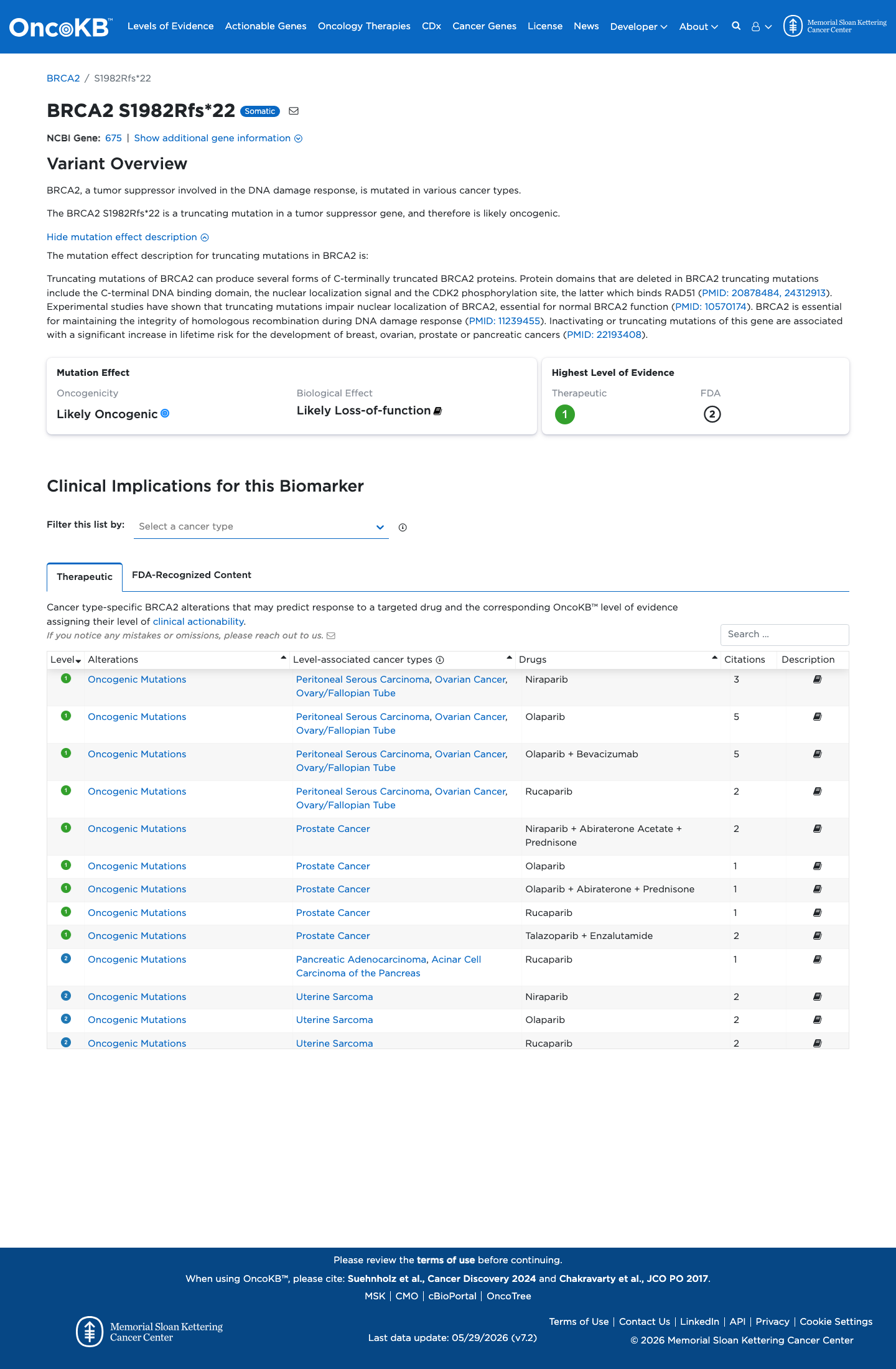
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
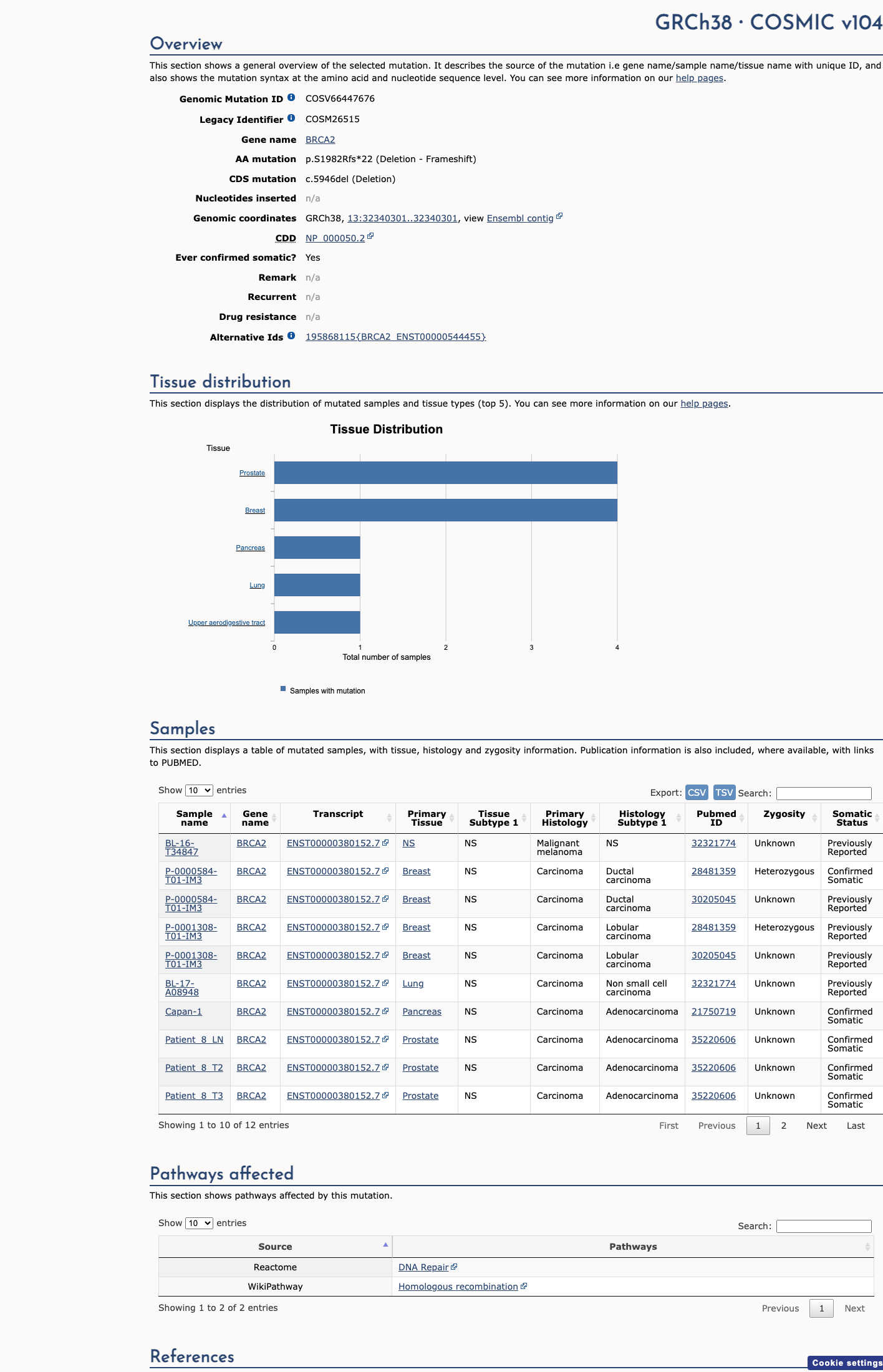
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV66447676, n = 10 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 9 further PMIDs triaged but not cited — see Sources & References.
Li et al. 2020 BRCA1/2 clinical-history likelihood-ratio model
Found
Structured finding pending for this record — see source link.
Applied to
→PP4 supports · met
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
20878484 ↗
A new mutation of BRCA2 gene in an Italian healthy woman with familial breast cancer history.
ONCOKB
24312913 ↗
A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer.
ONCOKB
10417300 ↗
De novo BRCA1 mutation in a patient with breast cancer and an inherited BRCA2 mutation.
CLINVAR
14559878 ↗
Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia.
CLINVAR
11466700 ↗
The frequency of founder mutations in the BRCA1, BRCA2, and APC genes in Australian Ashkenazi Jews: implications for the generality of U.S. population data.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR