NM_000051.4:c.7381C>T (p.Arg2461Cys) is a missense variant in ATM, a gene where loss of function is an established mechanism for Ataxia-Telangiectasia (AR) and heterozygous variants confer moderate risk for breast and other cancers (AD). This variant is not a null variant and SpliceAI predicts no splicing impact (max delta 0.07); PVS1 is not applicable per the ATM VCEP v1.5.0 decision tree.1 This variant is present in gnomAD v4.1 at an overall allele frequency of 0.00613% (99/1,614,160 alleles; 0 homozygotes) with a grpmax filtering AF of 0.039%. The frequency exceeds the PM2_Supporting threshold (≤0.001%) but does not reach BS1 (>0.05%) or BA1 (>0.5%).2 In silico predictions are inconclusive: REVEL score of 0.707 falls between the PP3 threshold (>0.7333) and BP4 missense threshold (≤0.249), neither meeting nor refuting pathogenicity. However, SpliceAI predicts no splicing impact (max delta 0.07 ≤ 0.1), satisfying BP4_Supporting.3 This variant has been reported in ClinVar (VCV000142541) with predominantly uncertain significance classifications (14 clinical laboratories), with a minority of likely benign (3) and benign (1) submissions. No expert panel classification is available.4 Functional evidence from the VCEP-validated Barone 2009 (PMID:19431188) kinase assay suggests partial impairment of ATM autophosphorylation, potentially supporting PS3_Supporting, but full-text verification of the variant-specific result was not possible in this assessment. Human review is recommended.5 No case-control study meeting PS4 criteria, no published segregation data for PP1, and no phased trans/cis data for PM3/BP2 were identified for this variant. Applying the ATM VCEP v1.5.0 criteria combination rules (Richards et al. 2015), only BP4_Supporting is met with no pathogenic criteria satisfied. The variant is classified as a Variant of Uncertain Significance (VUS). If PS3_Supporting is confirmed upon human review of the functional data, the variant would remain VUS (1 pathogenic supporting + 1 benign supporting under Rule31).6
ATM
Final classification
VUS
ATM c.7381C>T · p.Arg2461Cys
ATM
NM_000051.4:c.7381C>T (p.Arg2461Cys) is a missense variant in ATM, a gene where loss of function is an established mechanism for Ataxia-Telangiectasia (AR) and heterozygous variants confer moderate risk for breast and other cancers (AD).
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework was evaluated deterministically with applied criteria: BP4 supporting; no rule matched the adjudicated criteria.
Classification rationale
BP4
VUS
ATM c.7381C>T
BP4
→
VUS
1
spliceai ↗vcep_atm_pvs1_1_5
3
revelspliceai ↗cspec ↗
5
vcep_clingen_hbop_atm_supplementary_tables_1_and_2_v1
6
cspec ↗
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 9 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BP4
supporting
Benign
ATM VCEP applies BP4_Supporting when SpliceAI predicts no splicing impact (max delta ≤0.1). SpliceAI predicts no significant splice impact for c.7381C>T (max delta score = 0.07), meeting the VCEP BP4 threshold for absence of predicted splicing disruption. The REVEL score of 0.707 does not meet the missense BP4 threshold (≤0.249), but BP4 is independently satisfied by the splicing computational evidence.
SpliceAI max delta 0.07 ≤ VCEP BP4 threshold of 0.1 — no predicted splicing impactREVEL 0.707 does not separately meet BP4 missense threshold of ≤0.249
Assessed · not applied
Pathogenic
PS3
The ATM VCEP functional assay spreadsheet (clingen_hbop_atm_supplementary_tables_1_and_2_v1.xlsx) validates Barone 2009 (PMID:19431188) and Mitui 2009 (PMID:18634022) kinase assays as approved for PS3_Supporting/BS3_Supporting.
PS4
ATM VCEP requires a case-control study with p≤0.05 and OR≥2 (or lower 95% CI≥1.5) for PS4_Strong.
PM2
ATM VCEP applies PM2_Supporting when frequency is ≤0.001% in gnomAD v4.
PP1
ATM VCEP applies PP1 for AR condition (Ataxia-Telangiectasia) when co-segregation is observed in affected relatives carrying both variants.
PP3
ATM VCEP applies PP3_Supporting for missense variants when REVEL >0.7333, or for splicing when SpliceAI ≥0.2.
Benign
BA1
ATM VCEP applies BA1 Stand-Alone when grpmax filtering AF exceeds 0.5% in gnomAD v4.
BS1
ATM VCEP applies BS1_Strong when grpmax filtering AF exceeds 0.05% in gnomAD v4.
BS3
ATM VCEP applies BS3 when a variant rescues ATM-specific functional features.
BP2
ATM VCEP applies BP2 when a variant is observed in cis with a pathogenic variant for A-T (recessive) or homozygous/trans in an unaffected individual ≥18 years with no evidence of A-T.
N/A · 15
PVS1 · PS1 · PS2 · PM1 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP5 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
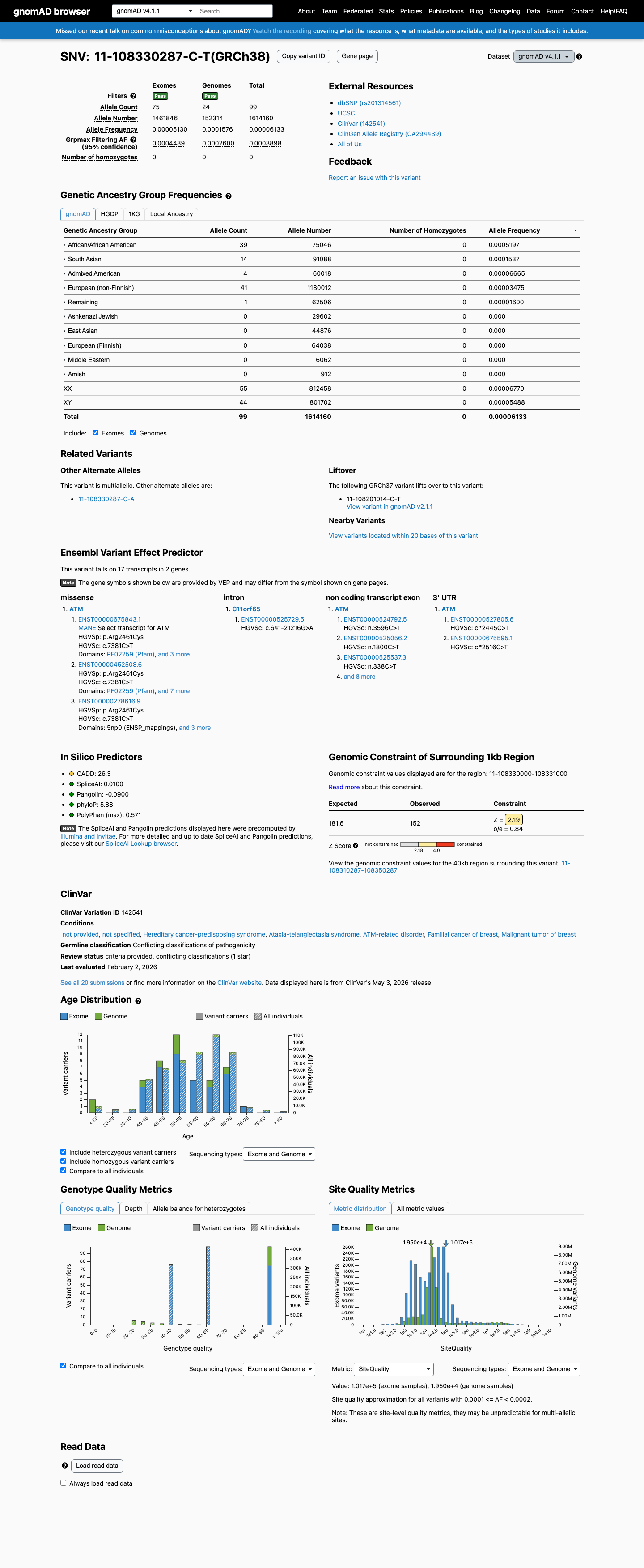
This variant is present in gnomAD v4.1 (AF= 6.13322e-05; MAF= 0.00613%, 99/1614160 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0.000519681; MAF= 0.05197%, 39/75046 alleles, homozygotes = 0); grpmax FAF= 0.00038979.
v2.1
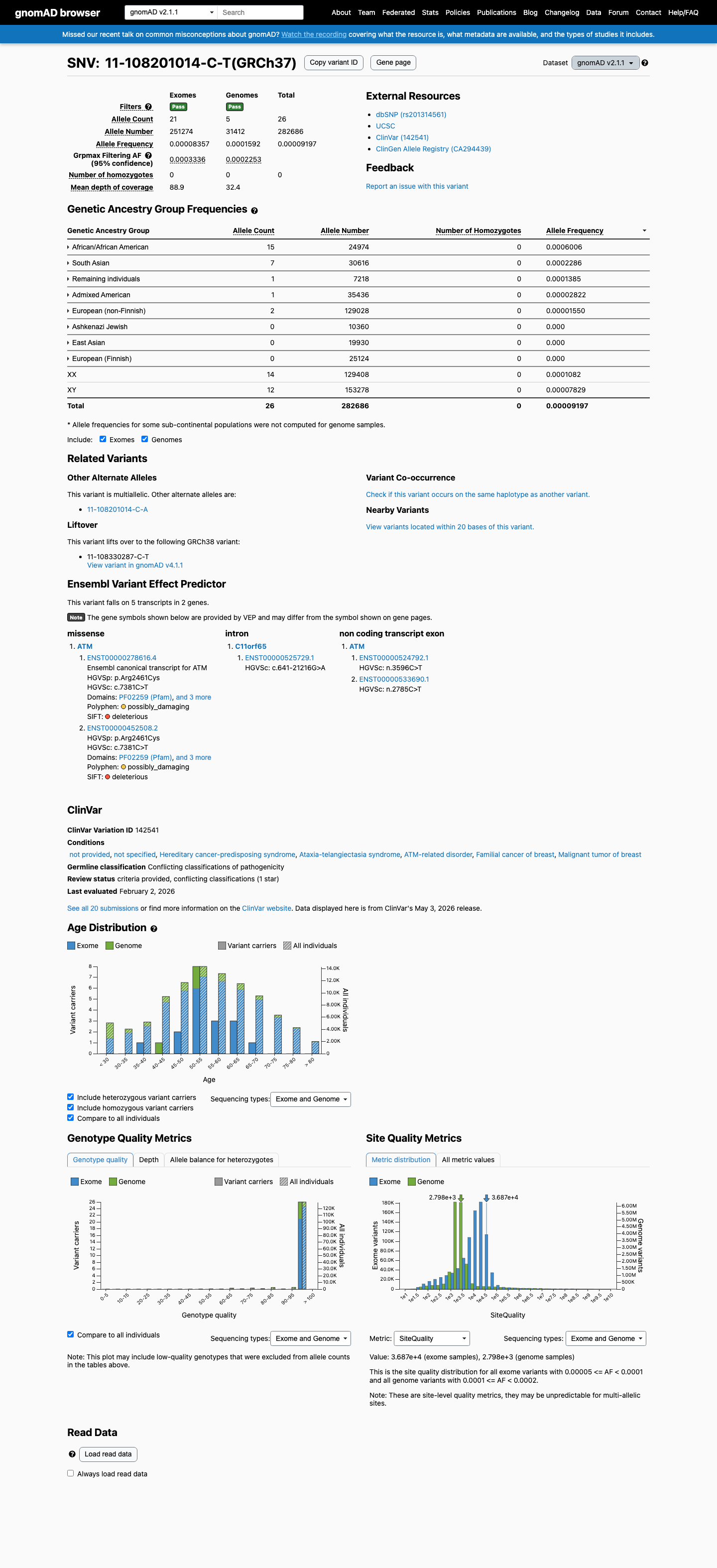
This variant is present in gnomAD v2.1 (AF= 9.19748e-05; MAF= 0.00920%, 26/282686 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0.000600625; MAF= 0.06006%, 15/24974 alleles, homozygotes = 0); grpmax FAF= 0.0003336.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0061%
· 99 / 1,614,160
0 hom · FAF 0.039%
0 hom · FAF 0.039%
African/African American 39 / 75,046 |
0.052% |
South Asian 14 / 91,088 |
0.015% |
Admixed American 4 / 60,018 |
0.0067% |
European (non-Finnish) 41 / 1,180,012 |
0.0035% |
Remaining individuals 1 / 62,506 |
0.0016% |
+ 5 not observed (European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0092%
· 26 / 282,686
0 hom · FAF 0.033%
0 hom · FAF 0.033%
African/African American 15 / 24,974 |
0.06% |
South Asian 7 / 30,616 |
0.023% |
Remaining individuals 1 / 7,218 |
0.014% |
Admixed American 1 / 35,436 |
0.0028% |
European (non-Finnish) 2 / 129,028 |
0.0016% |
+ 3 not observed (Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
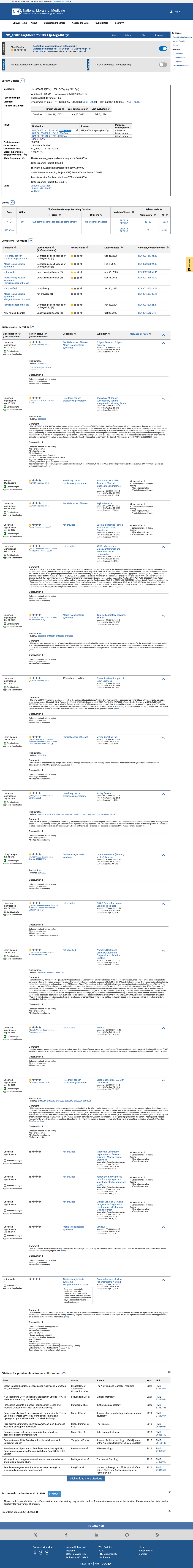
This variant has been reported in ClinVar as Uncertain significance (14 clinical laboratories) and as Likely benign (3 clinical laboratories) and as Benign (1 clinical laboratory) and as Uncertain Significance (1 clinical laboratory). (ClinVarID = 142541)
In silico
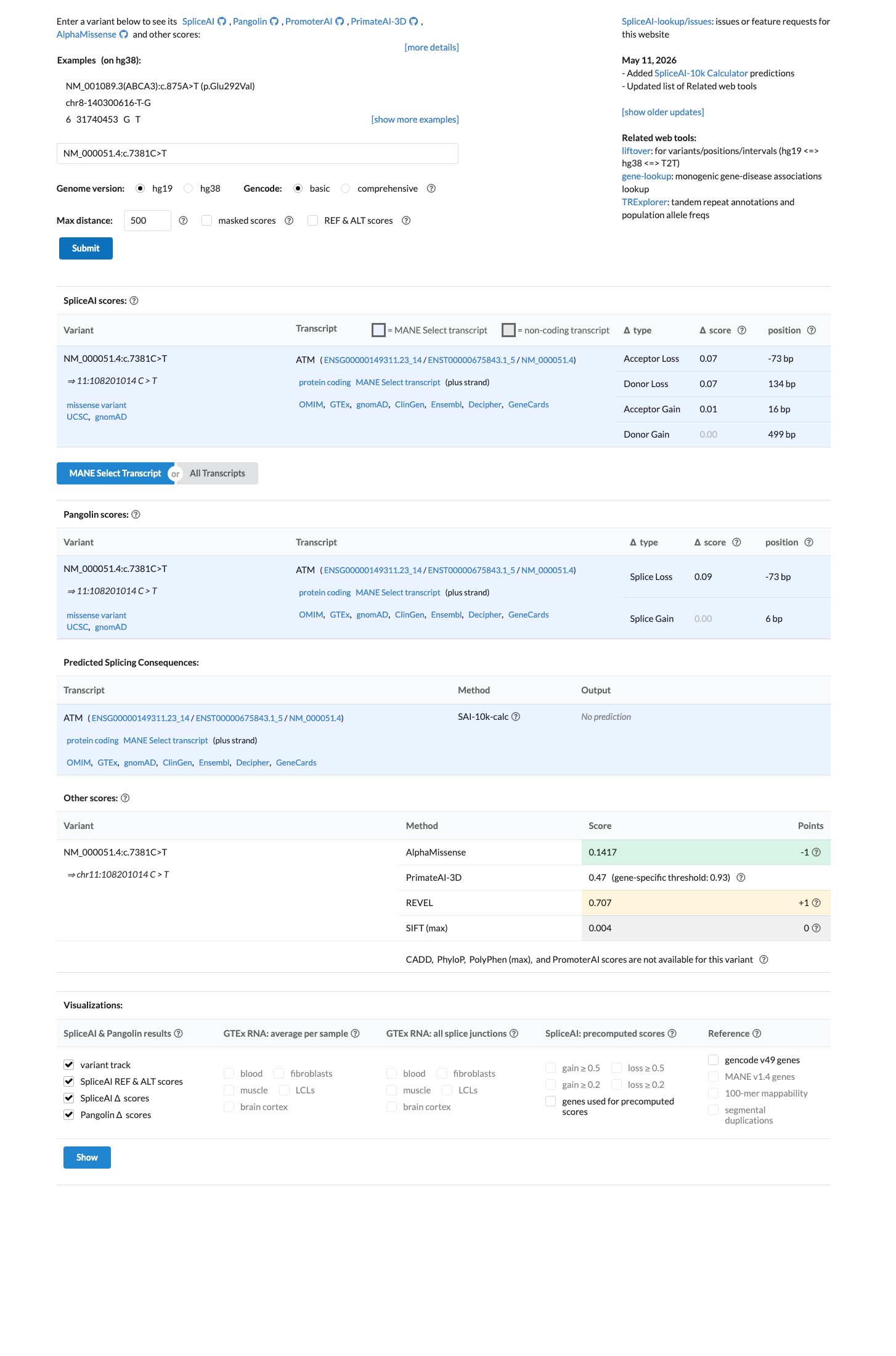
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.07). REVEL score = 0.707. BayesDel score = 0.109334.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. ATM, a kinase involved in the DNA damage response, is mutated in various solid and hematologic malignancies.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53748241, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 10 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26010451 ↗
Amplicon sequencing of colorectal cancer: variant calling in frozen and formalin-fixed samples.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
27498913 ↗
Monogenic and polygenic determinants of sarcoma risk: an international genetic study.
CLINVAR
27978560 ↗
Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer.
CLINVAR
29058119 ↗
Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR