NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) is a nonsense variant in exon 6 of 15 exons in CTNNB1, predicted to result in premature protein termination and nonsense-mediated decay of the transcript.1 CTNNB1 loss-of-function is an established disease mechanism for autosomal dominant neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV; MIM 615075), supported by ClinGen haploinsufficiency score of 3 and multiple publications describing pathogenic germline nonsense and frameshift variants.2 This variant is absent from gnomAD v2.1 and v4.1 (0 alleles across >140,000 individuals) and absent from gnomAD-Canada v1.0, consistent with a rare pathogenic variant under strong purifying selection.3 The variant is not present in ClinVar, and no functional studies, cosegregation data, or detailed clinical phenotypes for individuals carrying this variant were available in the evidence record. Applying the generic ACMG/AMP 2015 classification framework (Richards et al. 2015, PMID:25741868), the combination of one very strong criterion (PVS1) and one moderate criterion (PM2) meets the threshold for Likely Pathogenic (1 Very Strong + 1 Moderate).4
CTNNB1
Final classification
Likely Pathogenic
CTNNB1 c.802G>T · p.Gly268Ter
CTNNB1
NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) is a nonsense variant in exon 6 of 15 exons in CTNNB1, predicted to result in premature protein termination and nonsense-mediated decay of the transcript.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 moderate; combination = 1 very strong + 1 moderate, which maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
CTNNB1 c.802G>T
PVS1 + PM2
→
Likely Pathogenic
Gene diagram
· NM_001904.4 · variants mapped to exon structure
CTNNB1
NM_001904.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 16 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_001904.4:c.802G>T is a nonsense variant in exon 6 of 15 exons, predicted to result in a premature termination codon (NP_001895.1:p.Gly268Ter) and trigger nonsense-mediated decay (stop codon >50 nt upstream of the last exon-exon junction). CTNNB1 loss-of-function is an established disease mechanism for autosomal dominant neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV; MIM 615075), supported by ClinGen haploinsufficiency score of 3 and multiple clinical publications describing germline nonsense and frameshift variants as pathogenic. Per the ClinGen SVI PVS1 decision tree (PMC6185798), a nonsense variant in a gene with an established LoF mechanism where NMD is predicted qualifies for PVS1 at very strong strength.
Nonsense variant NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) in exon 6 of 15 exons>50 nt upstream of last exon-exon junction — NMD predictedCTNNB1 loss-of-function is an established germline disease mechanism for autosomal dominant NEDSDV (MIM 615075)
✓
PM2
moderate
Pathogenic
NM_001904.4:c.802G>T is completely absent from large population databases. It is absent from gnomAD v2.1 (0 alleles), gnomAD v4.1 (0 alleles across >140,000 individuals), and gnomAD-Canada v1.0 (0 alleles). Under generic ACMG/AMP non-VCEP cutoffs, absence from population databases supports PM2 at moderate strength.
Absent from gnomAD v2.1 (0 alleles across all populations)Absent from gnomAD v4.1 (0 alleles across >140000 individuals)
Assessed · not applied
Pathogenic
PS2
The exploratory evidence pipeline identified a potential confirmed de novo report for this variant (PMID:32222192, Li et al.
PS3
No well-established functional studies (in vitro or in vivo) of NM_001904.4:c.802G>T are available in the evidence record.
PS4
The variant is absent from ClinVar, so affected individuals cannot be tallied from that source.
PM1
The variant does not lie in a known mutational hotspot or critical functional domain.
PM6
The exploratory evidence pipeline identified a possible assumed de novo record for this variant in DECIPHER, but parentage confirmation status could not be verified.
PP1
No cosegregation data is available.
PP3
Multiple lines of computational evidence supporting a deleterious effect are not available.
PP4
No detailed clinical phenotype information for individuals carrying NM_001904.4:c.802G>T was available for review.
PP5
No ClinVar entry exists for NM_001904.4:c.802G>T.
Benign
BA1
BA1 requires an allele frequency >5% in any general population.
BS1
BS1 requires an allele frequency greater than expected for the disorder (>0.3% under generic non-VCEP cutoffs).
BS2
BS2 requires observation in a healthy adult individual for a fully penetrant dominant disorder.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect on protein function or splicing are available for NM_001904.4:c.802G>T.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on the gene product.
BP5
No ClinVar entry exists for NM_001904.4:c.802G>T, and no verified publication reports a benign classification for this variant from a reputable source.
BP6
No ClinVar entry exists for NM_001904.4:c.802G>T with a benign classification from a reputable source.
N/A · 10
PS1 · PM3 · PM4 · PM5 · PP2 · BS4 · BP1 · BP2 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
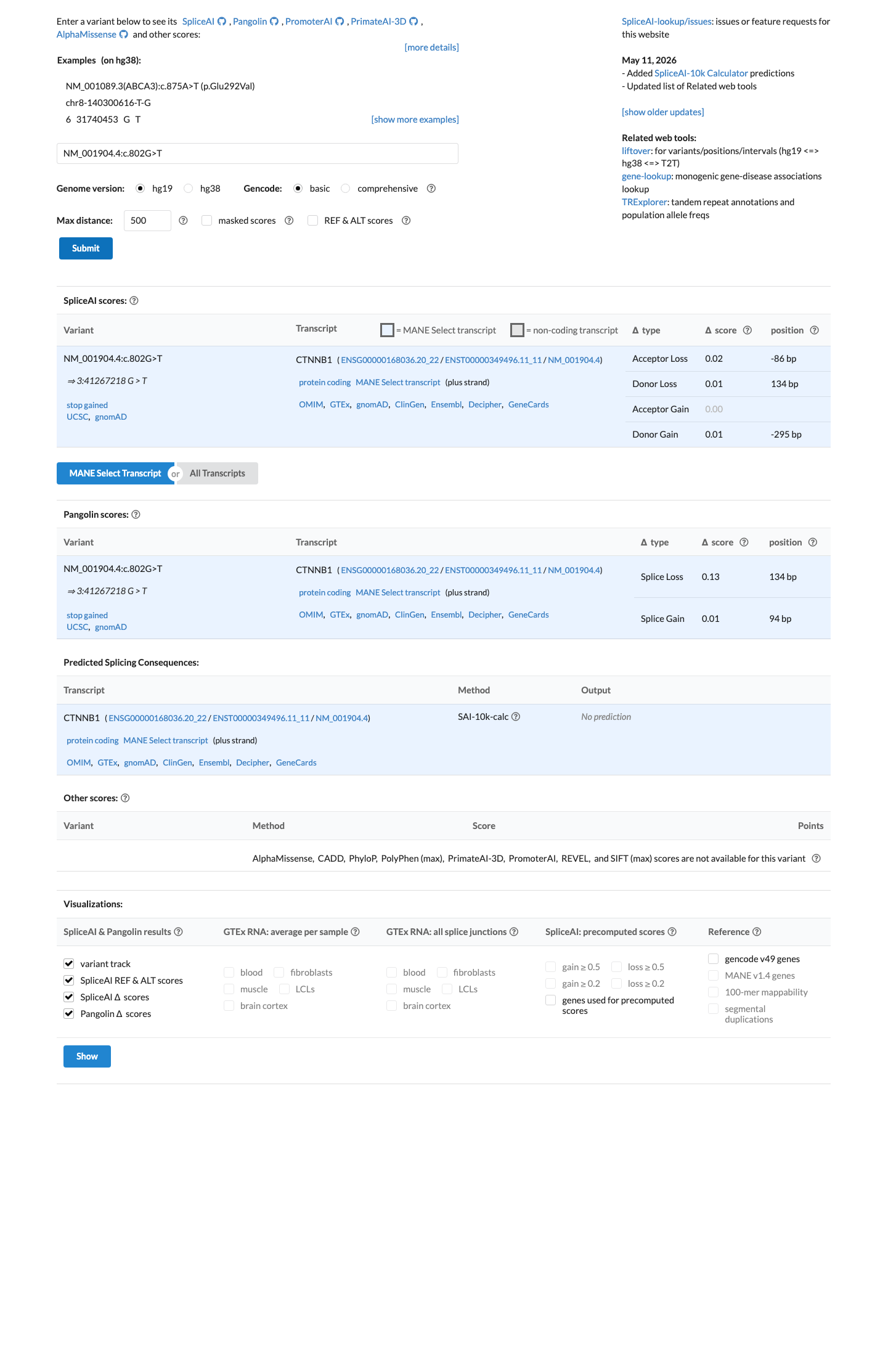
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). BayesDel score = 0.66.
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
5papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
PMID 33350591
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
PMID 36083290
Found
Nonsense variant NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) in exon 6 of 15 exons >50 nt upstream of last exon-exon junction — NMD predicted CTNNB1 loss-of-function is an established germline disease mechanism for autosomal dominant NEDSDV (MIM 615075) supported by ClinGen haploinsufficiency score of 3 Multiple publications confirm germline CTNNB1 nonsense/frameshift variants as pathogenic (PMID:41082542 PMID:36153650 PMID:36083290) ClinGen SVI PVS1 framework (PMC6185798) supports PVS1 at very strong for nonsense variants in LoF-established genes with predicted NMD
Applied to
→PVS1 supports · met
PMID 36153650
Found
Nonsense variant NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) in exon 6 of 15 exons >50 nt upstream of last exon-exon junction — NMD predicted CTNNB1 loss-of-function is an established germline disease mechanism for autosomal dominant NEDSDV (MIM 615075) supported by ClinGen haploinsufficiency score of 3 Multiple publications confirm germline CTNNB1 nonsense/frameshift variants as pathogenic (PMID:41082542 PMID:36153650 PMID:36083290) ClinGen SVI PVS1 framework (PMC6185798) supports PVS1 at very strong for nonsense variants in LoF-established genes with predicted NMD
Applied to
→PVS1 supports · met
PMID 41062690
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
PMID 41082542
Found
Nonsense variant NM_001904.4:c.802G>T (NP_001895.1:p.Gly268Ter) in exon 6 of 15 exons >50 nt upstream of last exon-exon junction — NMD predicted CTNNB1 loss-of-function is an established germline disease mechanism for autosomal dominant NEDSDV (MIM 615075) supported by ClinGen haploinsufficiency score of 3 Multiple publications confirm germline CTNNB1 nonsense/frameshift variants as pathogenic (PMID:41082542 PMID:36153650 PMID:36083290) ClinGen SVI PVS1 framework (PMC6185798) supports PVS1 at very strong for nonsense variants in LoF-established genes with predicted NMD
Applied to
→PVS1 supports · met
Sources & reference links