NM_000051.4:c.5983G>T (p.Glu1995Ter) is a nonsense variant in exon 40 of 63 of ATM, a gene where loss of function is a well-established mechanism for autosomal recessive ataxia-telangiectasia. The premature termination codon lies well upstream of the final exon and is predicted to trigger nonsense-mediated decay, removing all critical C-terminal domains (FAT, PI3K, FATC). Under the ATM VCEP v1.5 PVS1 decision tree, this meets PVS1 at Very Strong strength.1 The variant is absent from gnomAD v4.1 and v2.1 (0 alleles observed), meeting the ATM VCEP v1.5 PM2_Supporting threshold of allele frequency ≤0.001% in the gnomAD v4 dataset.2 As a truncating variant with a premature termination codon at p.Glu1995, located upstream of p.Arg3047, PM5_Supporting applies per ATM VCEP v1.5 specifications.3 No experimental functional data (PS3/BS3), case-control enrichment data (PS4), de novo observations (PS5), or co-segregation data (PP1) were identified for this specific variant in the reviewed literature. None of the three publications reviewed (PMID:27413114, PMID:30348496, PMID:30553448) mentioned NM_000051.4:c.5983G>T. Applying the ATM VCEP v1.5 final classification rules (Richards et al. 2015 combination framework): PVS1 (Very Strong) plus two Supporting pathogenic criteria (PM2_Supporting, PM5_Supporting) satisfies Rule 4 (1 Pathogenic Very Strong + ≥2 Pathogenic Supporting), resulting in a final classification of Pathogenic.4
ATM
Final classification
Pathogenic
ATM c.5983G>T · p.Glu1995Ter
ATM
NM_000051.4:c.5983G>T (p.Glu1995Ter) is a nonsense variant in exon 40 of 63 of ATM, a gene where loss of function is a well-established mechanism for autosomal recessive ataxia-telangiectasia. The premature termination codon lies well upstream of the final exon and is predicted to trigger nonsense-mediated decay, removing all critical C-terminal domains (FAT, PI3K, FATC). Under the ATM VCEP v1.5 PVS1 decision tree, this meets PVS1 at Very Strong strength.
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework: matched Rule4 (1 Pathogenic.Very Strong + Pathogenic.Supporting >=2) with applied criteria: PVS1 very strong, PM2 supporting, PM5 supporting; maps to Pathogenic.
Classification rationale
PVS1PM2PM5
Pathogenic
ATM c.5983G>T
PVS1 + PM2 + PM5
→
Pathogenic
1
vcep_atm_pvs1_1_5vcep_suppl_tables1_pmid_40580951pvs1_generic_framework ↗
3
cspec ↗
4
cspec ↗final_classification_framework
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 7 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_000051.4:c.5983G>T is a nonsense variant (p.Glu1995Ter) in exon 40 of 63 of ATM, a gene where loss of function is a well-established disease mechanism for ataxia-telangiectasia. The premature termination codon lies well upstream of the final exon-exon junction and is predicted to trigger nonsense-mediated decay. The ATM VCEP v1.5 PVS1 decision tree was applied; the variant truncates the protein prior to the FAT domain (aa 2097–2488), PI3K domain (aa 2623–2974), and FATC domain (aa 3026–3055). The VCEP supplement (PMID:40580951, Table S1) classifies this variant as 'Non-functional' with a combined score of 0.960.
Nonsense variant at codon 1995 (E1995*) in exon 40 of 63 exonsPredicted to trigger nonsense-mediated decayTruncation upstream of all critical C-terminal domains (FAT
✓
PM2
supporting
Pathogenic
NM_000051.4:c.5983G>T is absent from gnomAD v4.1 (0 alleles out of population database) and gnomAD v2.1. The allele frequency of 0.0% is well below the ATM VCEP v1.5 PM2_Supporting threshold of ≤0.001% in the gnomAD v4 dataset.
Absent from gnomAD v4.1 (AF = 0)Absent from gnomAD v2.1 (AF = 0)Frequency ≤0.001% per VCEP PM2_Supporting threshold
✓
PM5
supporting
Pathogenic
NM_000051.4:c.5983G>T is a nonsense variant producing a premature termination codon at p.Glu1995Ter, which is upstream of p.Arg3047. Under ATM VCEP v1.5, PM5_Supporting applies to frameshifting or truncating variants with premature termination codons upstream of p.Arg3047.
Nonsense variant at p.Glu1995Terupstream of the PM5 cutoff residue p.Arg3047ATM VCEP v1.5 PM5_Supporting rule for truncating variants upstream of p.Arg3047
Assessed · not applied
Pathogenic
PS3
No experimental functional assay data were identified for NM_000051.4:c.5983G>T.
PS4
No case-control study meeting ATM VCEP v1.5 PS4 criteria (p ≤ 0.05 AND odds ratio/hazard ratio/relative risk ≥2 OR lower 95% CI ≥1.5) was identified for this variant.
PP1
No co-segregation data were identified for NM_000051.4:c.5983G>T in families with ataxia-telangiectasia.
Benign
BA1
NM_000051.4:c.5983G>T is absent from gnomAD v4.1.
BS1
NM_000051.4:c.5983G>T is absent from gnomAD v4.1.
BS3
No experimental functional assay data demonstrating rescue of ATM function were identified for NM_000051.4:c.5983G>T.
BP2
The ATM VCEP v1.5 BP2 point system requires observation of the variant in unaffected (non-A-T) individuals, scored by zygosity and phase relative to a pathogenic variant.
N/A · 18
PS1 · PS2 · PM1 · PM3 · PM4 · PM6 · PP2 · PP3 · PP4 · PP5 · BS2 · BS4 · BP1 · BP3 · BP4 · BP5 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant is present in ClinVar (Variation ID: 4077029); submission details unavailable.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.634849.
Functional
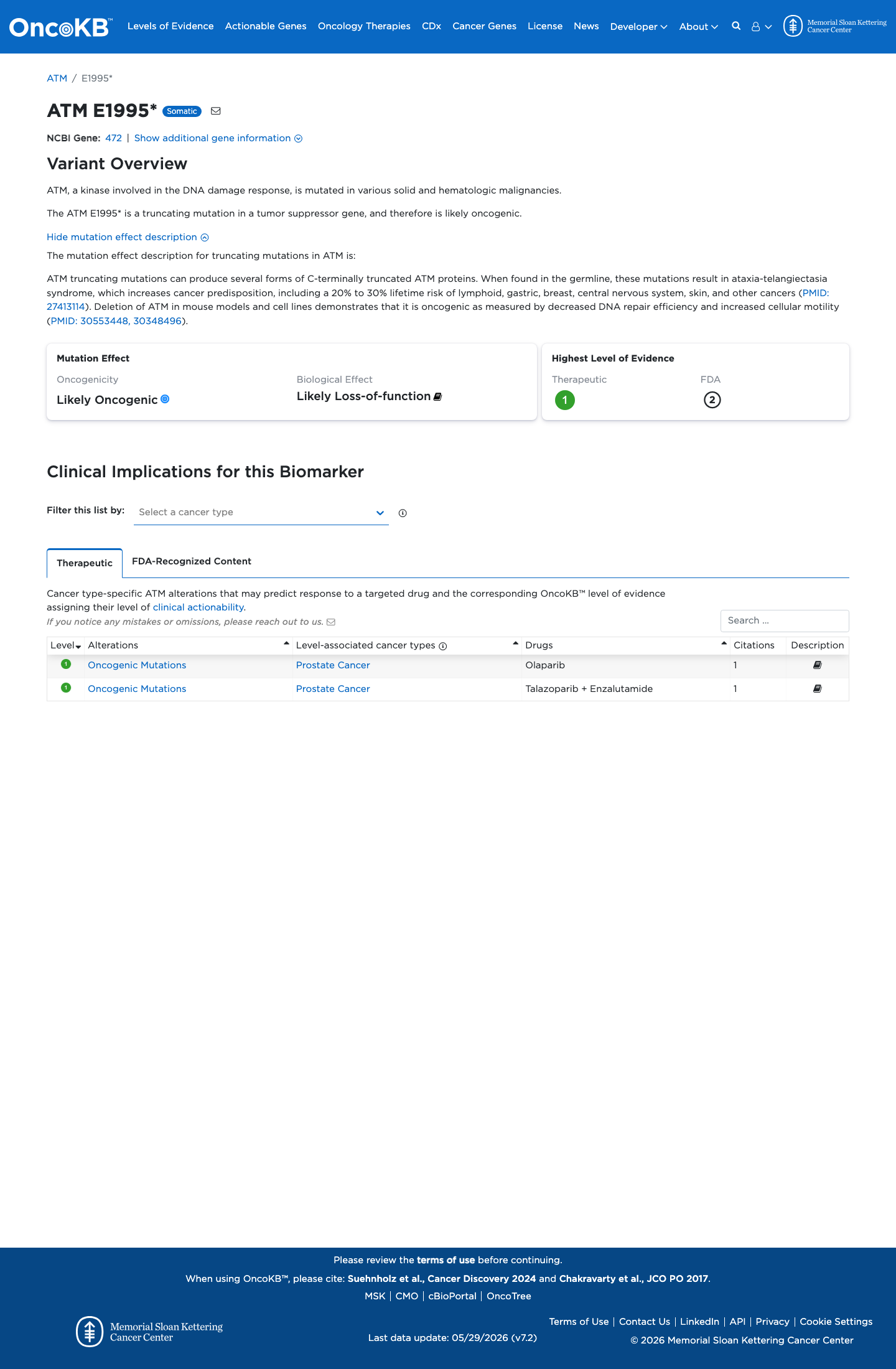
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
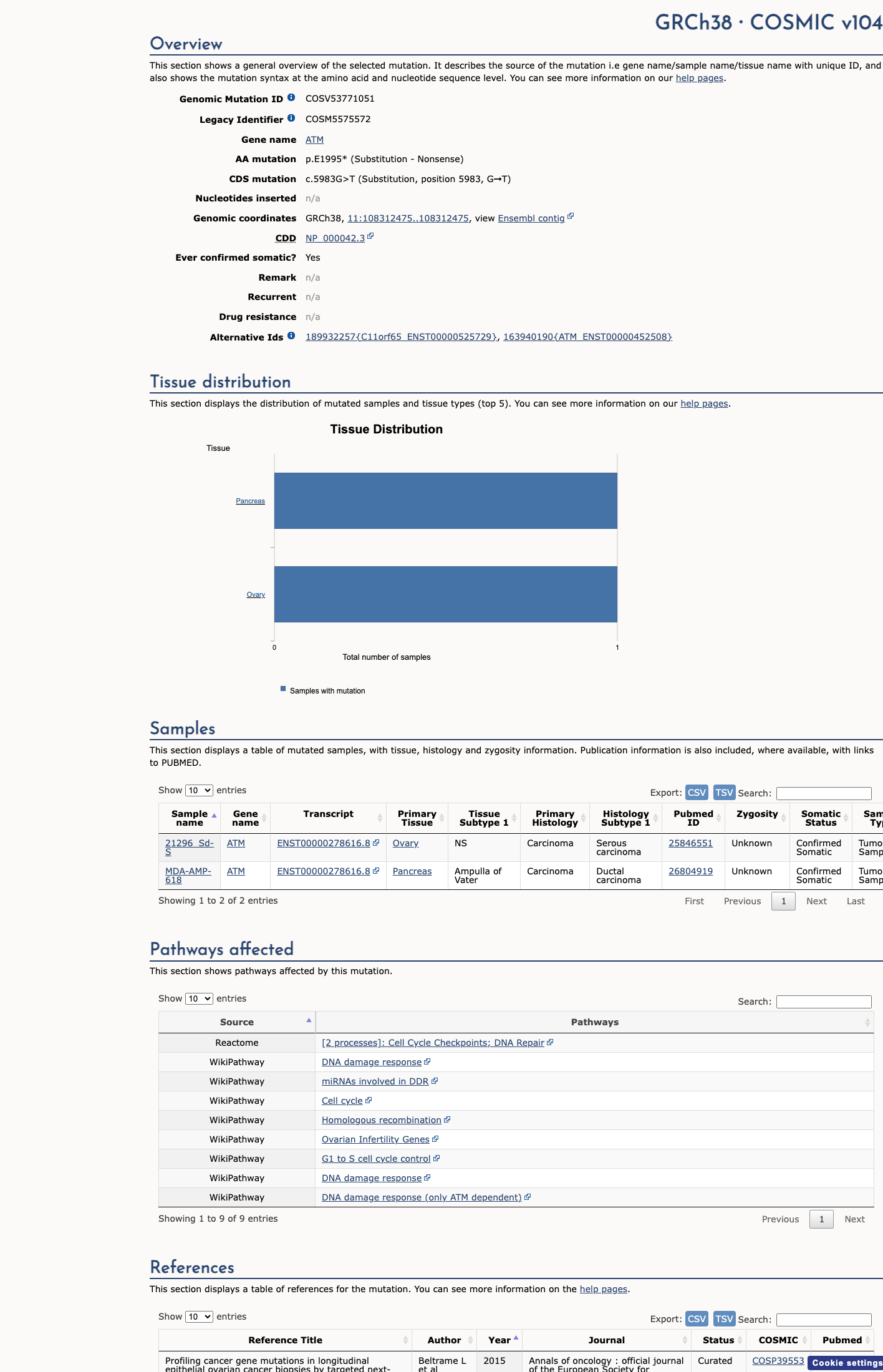
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53771051, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
30348496 ↗
Inactive Atm abrogates DSB repair in mouse cerebellum more than does Atm loss, without causing a neurological phenotype.
ONCOKB
30553448 ↗
Loss of ATM positively regulates Rac1 activity and cellular migration through oxidative stress.
ONCOKB