NM_000314.8:c.210-2A>G is a canonical splice acceptor site variant (AG>GG at the -2 position of intron 3) in PTEN, a gene where loss of function is an established mechanism for PTEN hamartoma tumor syndrome (PHTS).1 Per the ClinGen PTEN VCEP v3.2.0 PVS1 decision tree, this variant qualifies for PVS1 (Very Strong): it disrupts a canonical GT-AG splice site at a position 5' to p.D375 (c.1121), is predicted to cause exon 4 skipping with frameshift (p.Ala72Thrfs*5) and nonsense-mediated decay, and affects a biologically-relevant transcript (NM_000314.8). The closely related variant c.210-1G>A at the same splice junction has been experimentally confirmed to cause exon 4 skipping (PMID:28677221).2 The variant is absent from gnomAD v2.1 and v4.1, meeting the PTEN VCEP threshold for PM2_Supporting (allele frequency <0.001%).3 No direct functional evidence (RNA assay, minigene, or splicing study) was identified for c.210-2A>G specifically. The Mighell et al. 2018 phosphatase activity assay (mmc2.xlsx) only covers missense variants and is not applicable to this splice variant.4 SpliceAI predicts a max delta score of 0.00, which is anomalous for a canonical splice acceptor variant and may represent a lookup artifact. SpliceAI predictions should be independently verified before relying on computational splicing evidence for this variant.5 ClinVar reports this variant as Likely pathogenic (2 clinical laboratories) and Pathogenic (1 clinical laboratory) under ClinVar ID 576440, with review status 'criteria provided, single submitter.' However, BP6 is not for use per PTEN VCEP, and ClinVar classifications alone are not used as independent evidence.6 This variant has been observed in somatic cancers (COSMIC COSV64302273, n=4), supporting its potential role in tumorigenesis, though somatic occurrence alone is not an ACMG/AMP criterion for germline classification. A systematic review of the five available full-text publications (PMIDs: 16199547, 28677221, 29758562, 9467011, 21194675) confirmed that none mention the exact variant NM_000314.8:c.210-2A>G. The most relevant paper (PMID:28677221) studied 34 PTEN intronic variants including c.210-1G>A at the same splice junction, but c.210-2A>G was not among the variants analyzed.7 Combined evidence: PVS1 (Very Strong) + PM2_Supporting. Under the PTEN VCEP combination rules v3.2.0, this specific combination (1 Very Strong + 1 Supporting without an intermediate Moderate criterion) does not match any defined pathogenic or likely pathogenic rule. Under the generic ACMG/AMP 2015 framework (PMID:25741868), 1 Very Strong + 1 Supporting likewise does not reach the threshold for Likely Pathogenic (requires >=2 Supporting or >=1 Moderate). Using a Bayesian point system (PVS1=8, PM2=1, total=9), this falls within the Likely Pathogenic range (6-9 points). A final classification should be determined by the interpreting clinical laboratory considering the strength of the PVS1 call and the anomalous SpliceAI result.8
PTEN
Final classification
Likely Pathogenic
PTEN c.210-2A>G · p.?
PTEN
NM_000314.8:c.210-2A>G is a canonical splice acceptor site variant (AG>GG at the -2 position of intron 3) in PTEN, a gene where loss of function is an established mechanism for PTEN hamartoma tumor syndrome (PHTS).
Richards et.al., 2015 - Combining rules v3.2.0 criteria-combination framework: matched Rule20 (1 Pathogenic.Very Strong + 1 Pathogenic.Supporting) with applied criteria: PVS1 very strong, PM2 supporting; maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
PTEN c.210-2A>G
PVS1 + PM2
→
Likely Pathogenic
1
cspec ↗pvs1_gene_context
2
vcep_pvs1_decisiontree_ptenpvs1_variant_assessmentPMID:28677221 ↗
4
vcep_mmc2
8
cspec ↗generic_acmg_combination_rules
Gene diagram
· NM_000314.8 · variants mapped to exon structure
PTEN
NM_000314.8
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
Population frequency
“
Absent from gnomAD v4.1.
“
Absent from gnomAD v2.1.
“
This variant is absent from gnomAD-Canada.
ClinVar
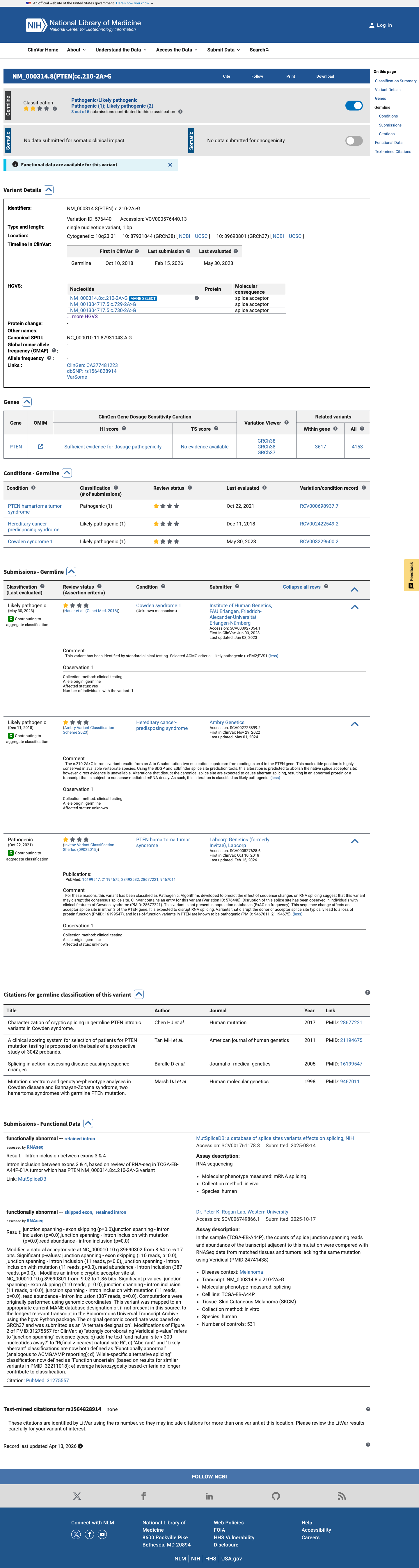
This variant has been reported in ClinVar as Likely pathogenic (2 clinical laboratories) and as Pathogenic (1 clinical laboratory). (ClinVarID = 576440)
In silico
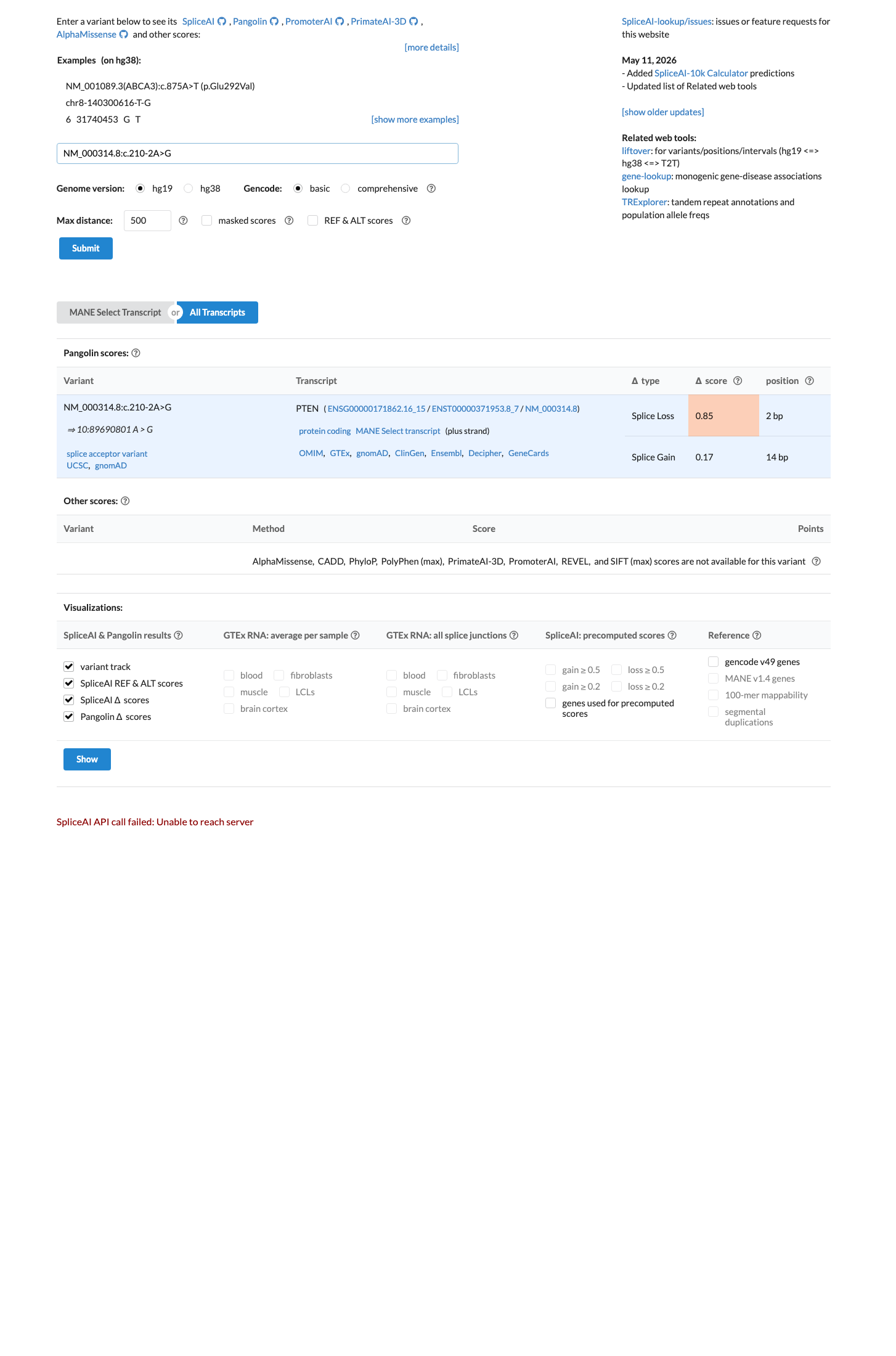
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.224139.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
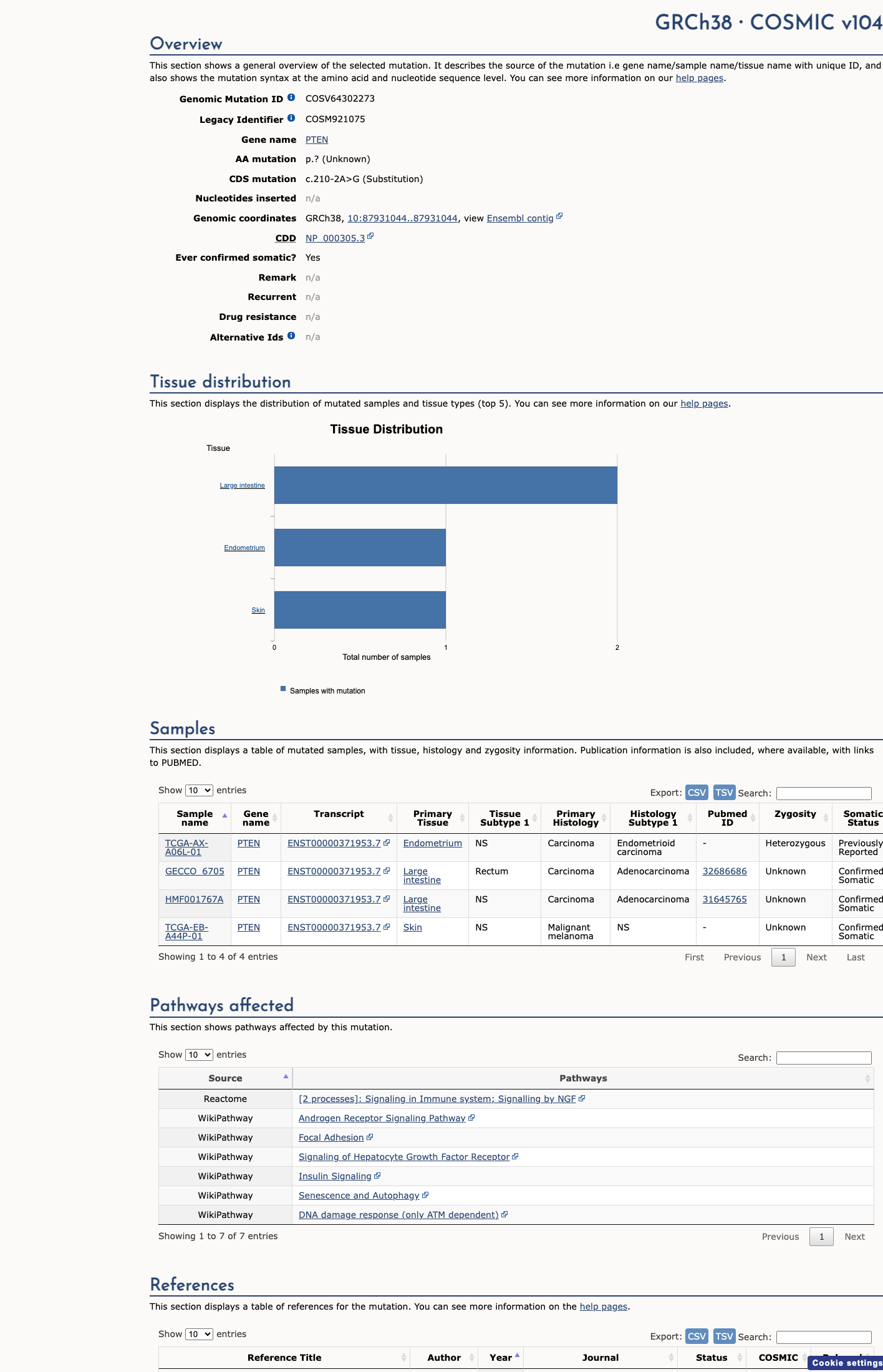
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV64302273, n = 4 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · 6 PMIDs triaged · 5 high-priority
6papers screened
Papers triaged by theme: functional/splicing/segregation/case_observation. high_priority_papers include abstract snippets. Use these to support PS3/BS3/PS4/PP1/PP3/PP5.
16199547 ↗
splicing rna
Splicing in action: assessing disease causing sequence changes.
Variations in new splicing regulatory elements are difficult to identify exclusively by sequence inspection and may result in deleterious effects on precursor (pre) mRNA splicing. These mutations can result in either complete skipping of the exon, retention of the intron, or the introduction of a new splice site within an exon or intron. Sometimes mutations that do not disrupt or create a splice s
BP7PP3PS3PS4PVS1
28677221 ↗
splicing rna
Characterization of cryptic splicing in germline PTEN intronic variants in Cowden syndrome.
Germline mutations in the tumor-suppressor gene PTEN predispose to subsets of Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome, and autism. Evidence-based classification of PTEN variants as either deleterious or benign is urgently needed for accurate molecular diagnosis and gene-informed genetic counseling. We studied 34 different germline PTEN intronic variants from 61 CS patients, charact
BP7PP3PS3PS4PVS1
29758562 ↗
functional
Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature.
PurposeShort stature is a common condition of great concern to patients and their families. Mostly genetic in origin, the underlying cause often remains elusive due to clinical and genetic heterogeneity.MethodsWe systematically phenotyped 565 patients where common nongenetic causes of short stature were excluded, selected 200 representative patients for whole-exome sequencing, and analyzed the ide
BS3PS3PS4
9467011 ↗
splicing rna
Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation.
The tumour suppressor gene PTEN , which maps to 10q23.3 and encodes a 403 amino acid dual specificity phosphatase (protein tyrosine phosphatase; PTPase), was shown recently to play a broad role in human malignancy. Somatic PTEN deletions and mutations were observed in sporadic breast, brain, prostate and kidney cancer cell lines and in several primary tumours such as endometrial carcinomas, malign
BP7PP3PS3PS4PVS1
21194675 ↗
case observation
A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands.
Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome are allelic, defined by germline PTEN mutations, and collectively referred to as PTEN hamartoma tumor syndrome. To date, there are no existing criteria based on large prospective patient cohorts to select patients for PTEN mutation testing. To address these issues, we conducted a multicenter prospective study in which 3042 probands satisfy
PS3PS4
28492532 ↗
background review
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
PS3PS4
Sources & reference links