Functional studies in transiently transfected mammalian cells demonstrate the p.Arg480Leu (R479L) mutant retains 75.5% activity toward 17-hydroxyprogesterone and 79.6% activity toward progesterone compared to wild-type, with normal substrate binding kinetics.1 The variant has been observed in a healthy population cohort at a frequency exceeding the benign threshold for a recessive disorder, with 1 heterozygote identified among 92 healthy Hungarian blood donors (1.09%).2 Multiple independent clinical diagnostic laboratories have classified this variant as Benign (three laboratories) or Likely benign (three laboratories) in ClinVar (variation ID 445854).3 The variant is also present in affected populations at a comparable low frequency (2/250 alleles, 0.8%, in a Sicilian CAH cohort), consistent with a benign polymorphism rather than a disease-causing allele.4 No pathogenic or likely pathogenic classifications for this variant have been reported by any clinical laboratory or expert panel.5
CYP21A2
Final classification
Likely Benign
CYP21A2 c.1439
CYP21A2
Functional studies in transiently transfected mammalian cells demonstrate the p.Arg480Leu (R479L) mutant retains 75.5% activity toward 17-hydroxyprogesterone and 79.6% activity toward progesterone compared to wild-type, with normal substrate binding kinetics.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 supporting, BS3 supporting, BP6 supporting; combination = 3 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BS3BP6
Likely Benign
CYP21A2 c.1439
BS1 + BS3 + BP6
→
Likely Benign
Applied criteria · 3 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
Population frequency
“
This variant is absent from gnomAD v4.1.
“
This variant is absent from gnomAD v2.1.
“
This variant is absent from gnomAD-Canada.
ClinVar
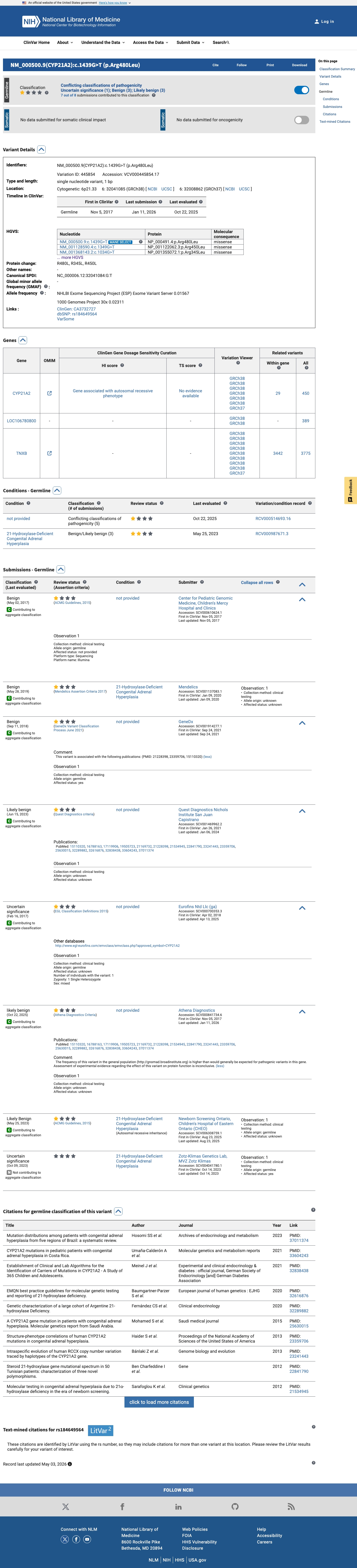
This variant has been reported in ClinVar as Benign (3 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Likely benign (1 clinical laboratory) and as likely benign (1 clinical laboratory) and as Likely Benign (1 clinical laboratory). (ClinVarID = 445854)
In silico
No data
No in-silico prediction was recorded for this variant.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Somatic evidence
COSMIC
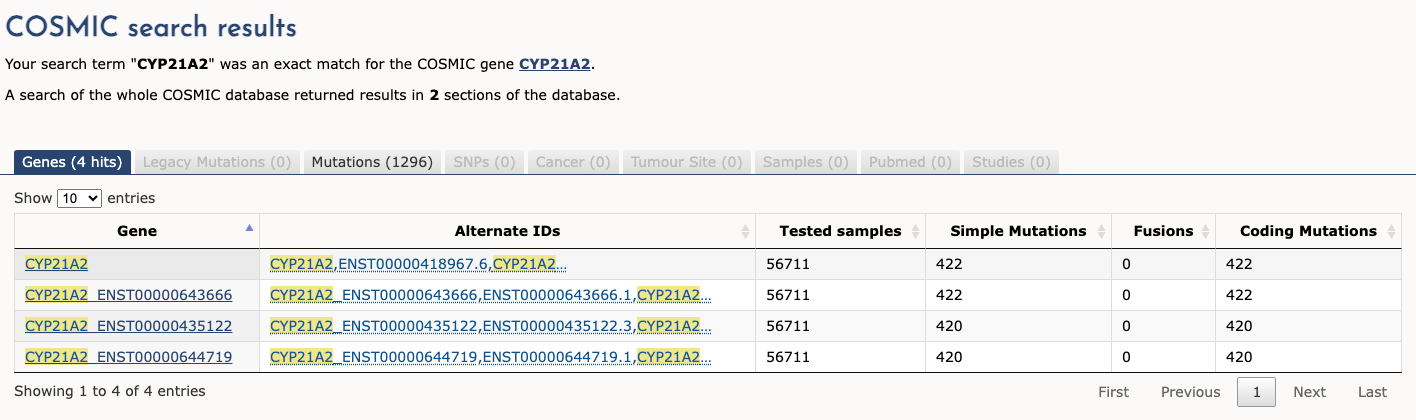
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · 20 PMIDs triaged · 8 high-priority
20papers screened
Papers triaged by theme: functional/splicing/segregation/case_observation. high_priority_papers include abstract snippets. Use these to support PS3/BS3/PS4/PP1/PP3/PP5.
15110320 ↗
functional
Detection and assignment of CYP21 mutations using peptide mass signature genotyping.
Congenital adrenal hyperplasia (CAH) is a common inborn error of steroidogenesis. The clinical spectrum of CAH ranges from the severe classical form, which can be fatal in the newborn, to simple virilizing forms or a milder non-classical form which is often not diagnosed until puberty. Recessive mutations in the autosomal gene encoding 21-hydroxylase (CYP21) are responsible for approximately 95% o
BS3PP5PS3PS4
16788163 ↗
functional
Molecular model of human CYP21 based on mammalian CYP2C5: structural features correlate with clinical severity of mutations causing congenital adrenal hyperplasia.
Enhanced understanding of structure-function relationships of human 21-hydroxylase, CYP21, is required to better understand the molecular causes of congenital adrenal hyperplasia. To this end, a structural model of human CYP21 was calculated based on the crystal structure of rabbit CYP2C5. All but two known allelic variants of missense type, a total of 60 disease-causing mutations and six normal v
BS3PP5PS3PS4
17119906 ↗
functional
Characterization of novel missense mutations in CYP21 causing congenital adrenal hyperplasia.
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is the most common inherited disorder of steroid metabolism, with an incidence of 1/10,000 in the general Caucasian population. Although most patients carry a deletion of the CYP21 gene or any of nine pseudogene-derived point mutations, the number of reported rare mutations continues to increase, and consist today of more than 80 diff
BS3PP5PS3PS4
19505723 ↗
splicing rna
Linkage analysis of the C4A/C4B copy number variation and polymorphisms of the adjacent steroid 21-hydroxylase gene in a healthy population.
Genes encoding the steroid 21-hydroxylase (CYP21A2) and the complement component C4 proteins (C4A and C4B) are located in the MHC region in a strongly linked structure named RCCX module. Previous studies found that carriers of C4B gene deficiency (C4B*Q0) have higher risk for cardiovascular diseases. A potential explanation is that lacking the C4B gene may result in altered function of the neighbo
BP7PP3PP5PS4PVS1
21169732 ↗
splicing rna
A large view of CYP21 locus among Sicilians and other populations: identification of a novel CYP21A2 variant in Sicily.
Several mutations in CYP21 locus cause 21-hydroxylase deficiency (21-OHD). The most common mutations are widespread among different geographic areas and their frequencies have been also reported to differ among certain populations. To obtain a large view on the frequencies of the most common mutations in the CYP21 locus, in Sicily, in the Mediterranean and other major geographic areas worldwide. T
BP7PP3PP5PS4PVS1
21228398 ↗
splicing rna
Carrier testing for severe childhood recessive diseases by next-generation sequencing.
Of 7028 disorders with suspected Mendelian inheritance, 1139 are recessive and have an established molecular basis. Although individually uncommon, Mendelian diseases collectively account for ~20% of infant mortality and ~10% of pediatric hospitalizations. Preconception screening, together with genetic counseling of carriers, has resulted in remarkable declines in the incidence of several severe r
BP7PP3PP5PS4PVS1
23241443 ↗
segregation
Intraspecific evolution of human RCCX copy number variation traced by haplotypes of the CYP21A2 gene.
The RCCX region is a complex, multiallelic, tandem copy number variation (CNV). Two complete genes, complement component 4 (C4) and steroid 21-hydroxylase (CYP21A2, formerly CYP21B), reside in its variable region. RCCX is prone to nonallelic homologous recombination (NAHR) such as unequal crossover, generating duplications and deletions of RCCX modules, and gene conversion. A series of allele-spec
BS4PP1PP5PS4
23359706 ↗
functional
Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia.
Mutations in the cytochrome p450 (CYP)21A2 gene, which encodes the enzyme steroid 21-hydroxylase, cause the majority of cases in congenital adrenal hyperplasia, an autosomal recessive disorder. To date, more than 100 CYP21A2 mutations have been reported. These mutations can be associated either with severe salt-wasting or simple virilizing phenotypes or with milder nonclassical phenotypes. Not all
BS3PP5PS3PS4
Sources & reference links