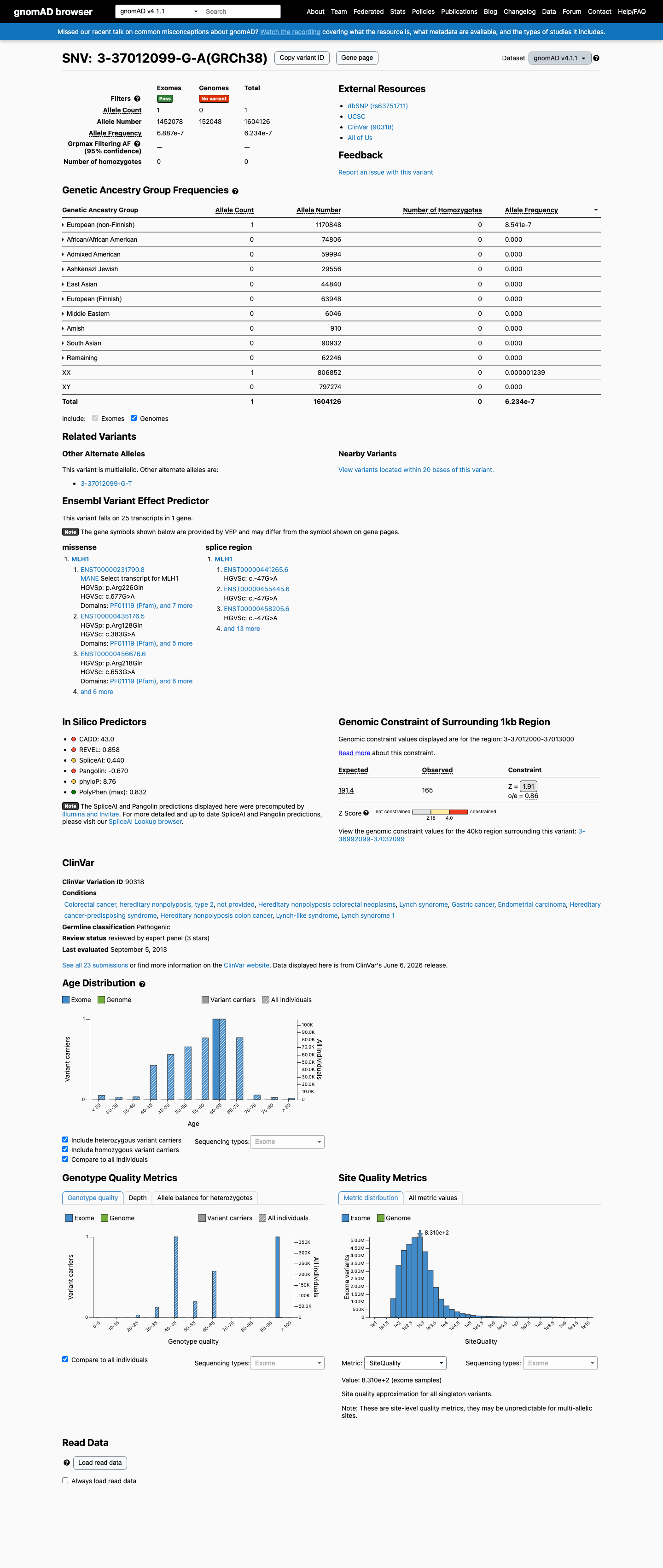
PVS1_VeryStrong: c.677G>A (last nucleotide of exon 8) produces no full-length transcript; patient mRNA analysis demonstrates complete exon 8 skipping (r.589_677del), frameshift, and NMD in two independent studies.1 PM2_Supporting: Extremely rare in population databases — gnomAD v4.1 AF = 6.23e-7 (1/1,604,126 alleles), absent from v2.1 and gnomAD-Canada, meeting VCEP threshold <0.00002.2 PP3_Supporting: SpliceAI predicts splicing impact (max delta = 0.44), meeting VCEP threshold (delta ≥0.2) for non-canonical splice position.3 PP4_Supporting: One CRC tumor from a carrier demonstrates MSI-H (5/5 markers) with loss of MLH1 protein expression by IHC.4 Evidence for pathogenicity includes confirmed RNA-level splicing aberration causing frameshift, extreme population rarity, in silico splice prediction, and tumor phenotype consistent with MMR deficiency.5
MLH1
Final classification
Pathogenic
MLH1 c.677G>A · p.Arg226Gln
MLH1
PVS1_VeryStrong: c.677G>A (last nucleotide of exon 8) produces no full-length transcript; patient mRNA analysis demonstrates complete exon 8 skipping (r.589_677del), frameshift, and NMD in two independent studies.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MLH1 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule4 (1 Pathogenic.Very Strong + Pathogenic.Supporting >=2) with applied criteria: PVS1 very strong, PM2 supporting, PP3 supporting, PP4 supporting, PP5 supporting; maps to Pathogenic.
Classification rationale
PVS1PM2PP3PP4PP5
Pathogenic
MLH1 c.677G>A
PVS1 + PM2 + PP3 + PP4 + PP5
→
Pathogenic
Gene diagram
· NM_000249.4 · variants mapped to exon structure
MLH1
NM_000249.4
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 11 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
c.677G>A (last nucleotide of exon 8) results in complete exon 8 skipping (r.589_677del) as demonstrated by patient mRNA analysis in two independent studies, leading to a frameshift and premature termination codon subject to nonsense-mediated decay. The variant allele produces no full-length or reference transcript. Confirmed in an affected sibling carrying the same variant.
Sharp et al. 2004: RT-PCR from patient lymphocytes shows c.677G>A causes complete skipping of exon 8 (89bp deletion)frameshiftand absence of mutant transcript from full-length product. Identical result in affected sister.
✓
PM2
supporting
Pathogenic
c.677G>A is present in gnomAD v4.1 at an allele frequency of 6.23e-7 (1/1,604,126 alleles, 0 homozygotes), which is below the VCEP PM2_Supporting threshold of <0.00002 (<1 in 50,000 alleles). The variant is absent from gnomAD v2.1 and gnomAD-Canada.
gnomAD v4.1: AF = 6.23e-71/1604
✓
PP3
supporting
Pathogenic
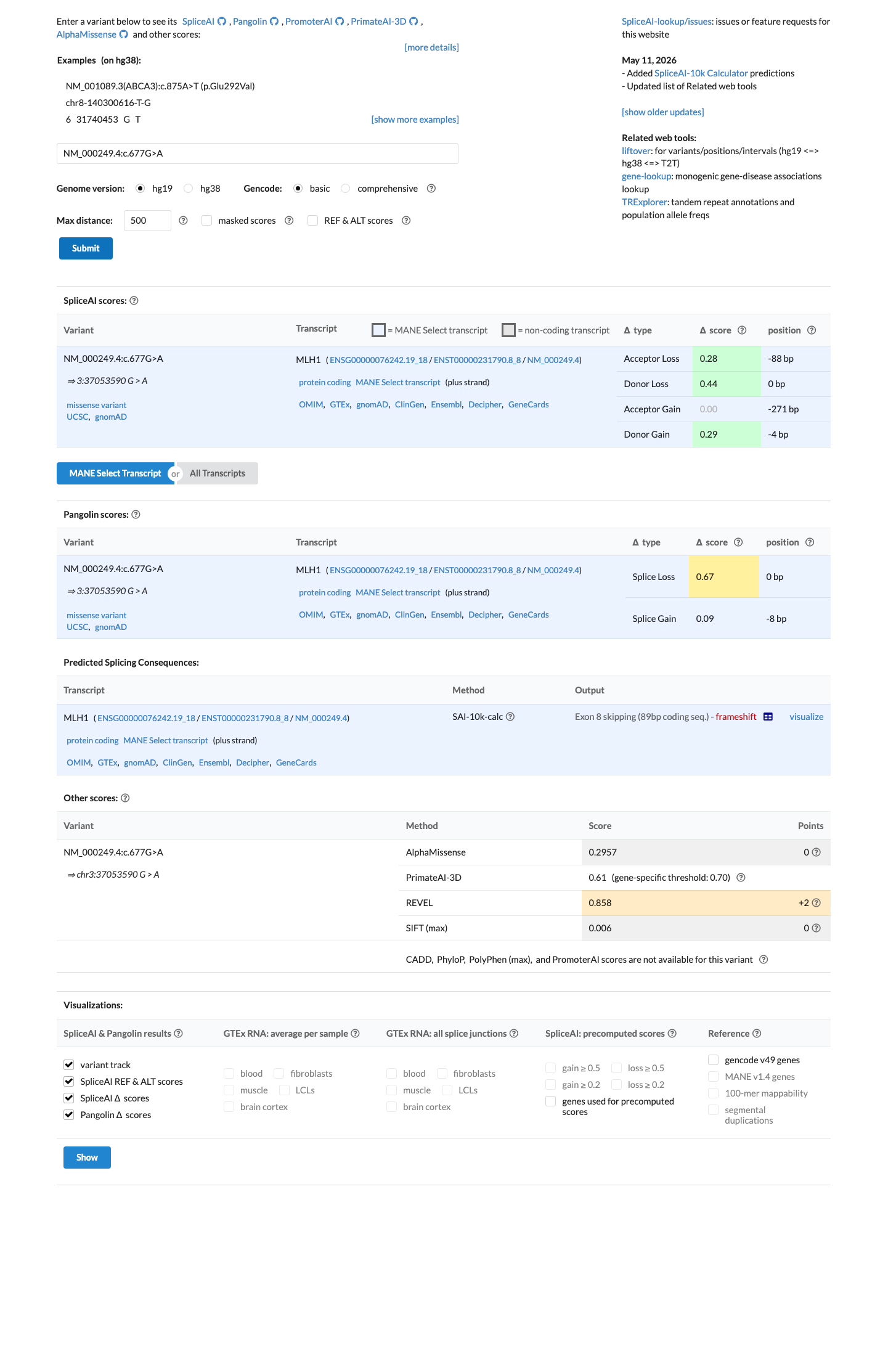
SpliceAI predicts a splicing defect for this variant (max delta score = 0.44), meeting the VCEP PP3_Supporting threshold of delta ≥0.2 for non-canonical splicing nucleotides. The HCI prior probability of 0.4702 does not meet the missense PP3 thresholds (>0.68).
SpliceAI: max delta = 0.44 (≥0.2 threshold for PP3_Supporting)HCI prior: 0.4702 (not ≥0.68 for missense PP3_Supportingnot >0.81 for PP3_Moderate)
✓
PP4
supporting
Pathogenic
One CRC tumor from a carrier of c.677G>A demonstrated MSI-H (5/5 microsatellite markers) and loss of MLH1 protein expression by immunohistochemistry (Pagenstecher et al. 2006). This meets VCEP PP4_Supporting criteria: 1 CRC with MSI-H and MMR protein loss consistent with variant location.
Pagenstecher et al. 2006Table 1patient 916: CRC at 31 years
Assessed · not applied
Pathogenic
PS2
No confirmed de novo occurrence of c.677G>A has been identified in ClinVar submissions, the published literature reviewed, or the InSiGHT LOVD database.
PS3
Variant was not found in the VCEP-calibrated functional assay documentation (Functional-assay-SVI-documentation-MMR.xlsx).
PP1
Pagenstecher et al.
Benign
BA1
gnomAD v4.1 allele frequency is 6.23e-7, well below the VCEP BA1 stand-alone benign threshold of ≥0.001 (0.1%).
BS1
gnomAD v4.1 allele frequency is 6.23e-7, below the VCEP BS1 strong benign threshold range of ≥0.0001 and <0.001.
BS2
No evidence of co-occurrence in trans with a known pathogenic MLH1 variant in a patient without clinical manifestations of CMMRD was identified in the reviewed literature.
BS3
All identified functional evidence demonstrates a damaging effect on splicing (complete exon 8 skipping).
BS4
Pagenstecher et al.
BP4
HCI prior probability of pathogenicity is 0.4702, far above the VCEP BP4_Supporting threshold of <0.11.
BP5
The available tumor evidence shows MSI-H with loss of MLH1 protein expression (Pagenstecher et al.
BP7
c.677G>A is a missense (non-synonymous) coding variant, not a synonymous or intronic variant.
N/A · 10
PS1 · PS4 · PM1 · PM5 · PM6 · PP2 · BP1 · BP2 · BP3 · BP6
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.23392e-07; MAF= 0.00006%, 1/1604126 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.54082e-07; MAF= 0.00009%, 1/1170848 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,604,126
0 hom
0 hom
European (non-Finnish) 1 / 1,170,848 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
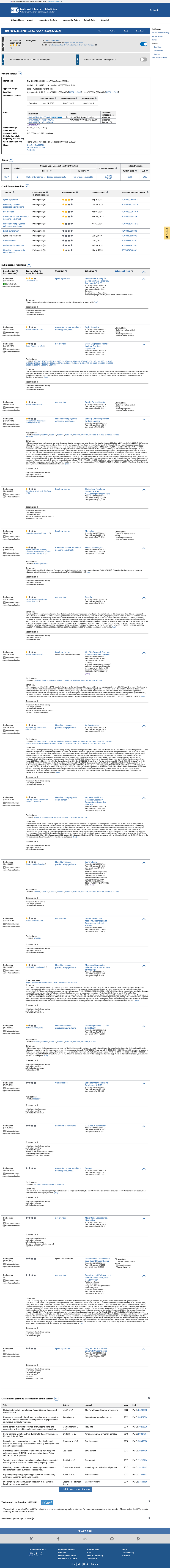
This variant has been reported in ClinVar as Pathogenic (18 clinical laboratories) and as Pathogenic by International Society for Gastrointestinal Hereditary Tumours (InSiGHT) (expert panel). (ClinVarID = 90318)
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 0.44). REVEL score = 0.198. BayesDel score = 0.384087. HCI prior probability for pathogenicity = 0.4702. MAPP score = 11.7. Custom PP2 score = 0.821.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MLH1, a DNA mismatch repair protein, is recurrently altered by deletion and mutation in various cancer types.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
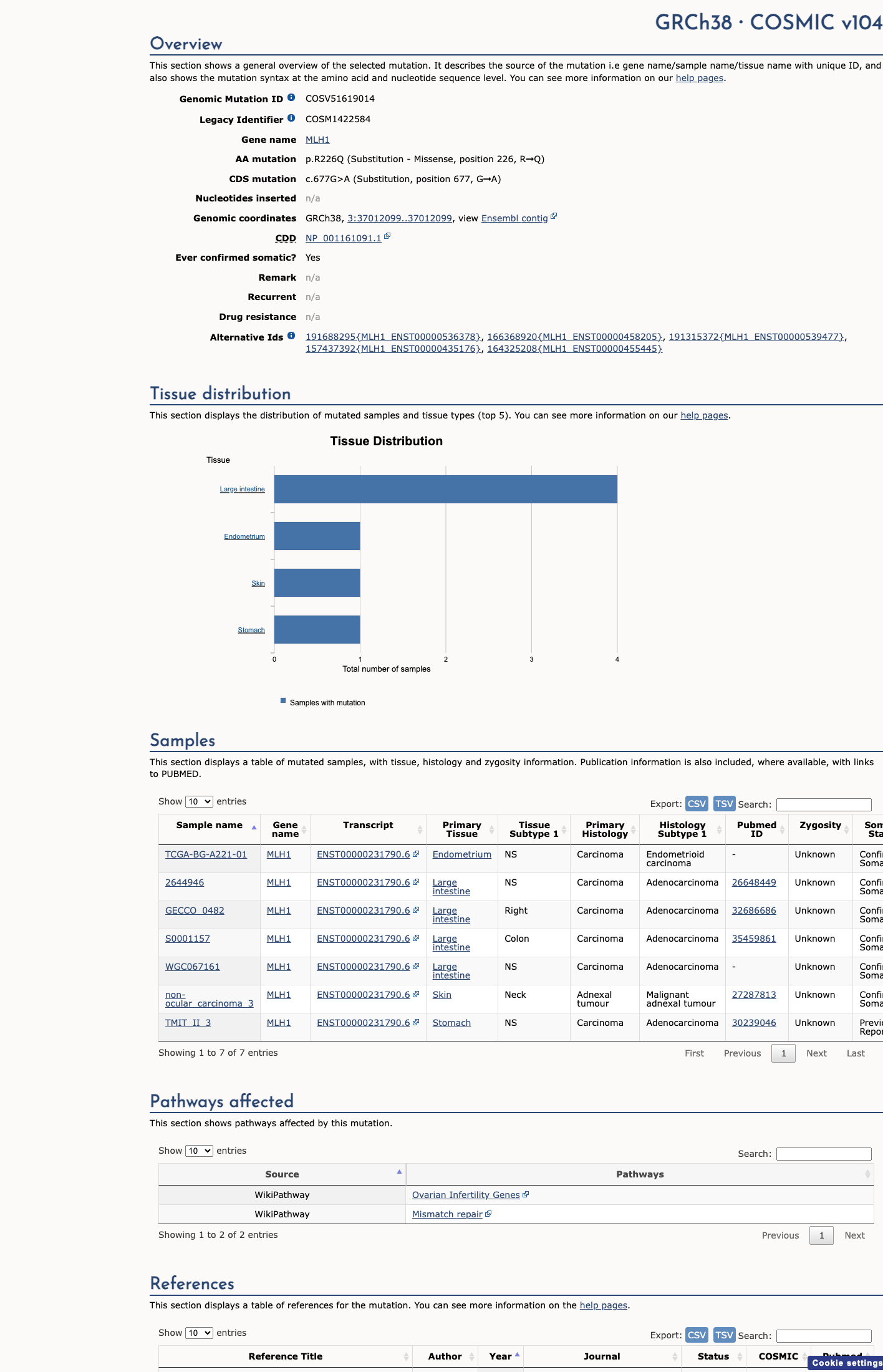
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV51619014, n = 7 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 5 further PMIDs triaged but not cited — see Sources & References.
Novel MLH1 and MSH2 germline mutations in the first HNPCC families identified in Slovakia.
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer.
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
RNA analysis reveals splicing mutations and loss of expression defects in MLH1 and BRCA1.
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants.
Found
Structured finding pending for this record — see source link.
Applied to
→PP4 supports · met
→PVS1 supports · met
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
15309712 ↗
Clinical features and mismatch repair gene mutation screening in Chinese patients with hereditary nonpolyposis colorectal carcinoma.
CLINVAR
16451135 ↗
Germline MSH2 and MLH1 mutational spectrum including large rearrangements in HNPCC families from Poland (update study).
CLINVAR
17453009 ↗
Patients with an unexplained microsatellite instable tumour have a low risk of familial cancer.
CLINVAR
17569143 ↗
Germline MLH1 and MSH2 mutational spectrum including frequent large genomic aberrations in Hungarian hereditary non-polyposis colorectal cancer families: implications for genetic testing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR