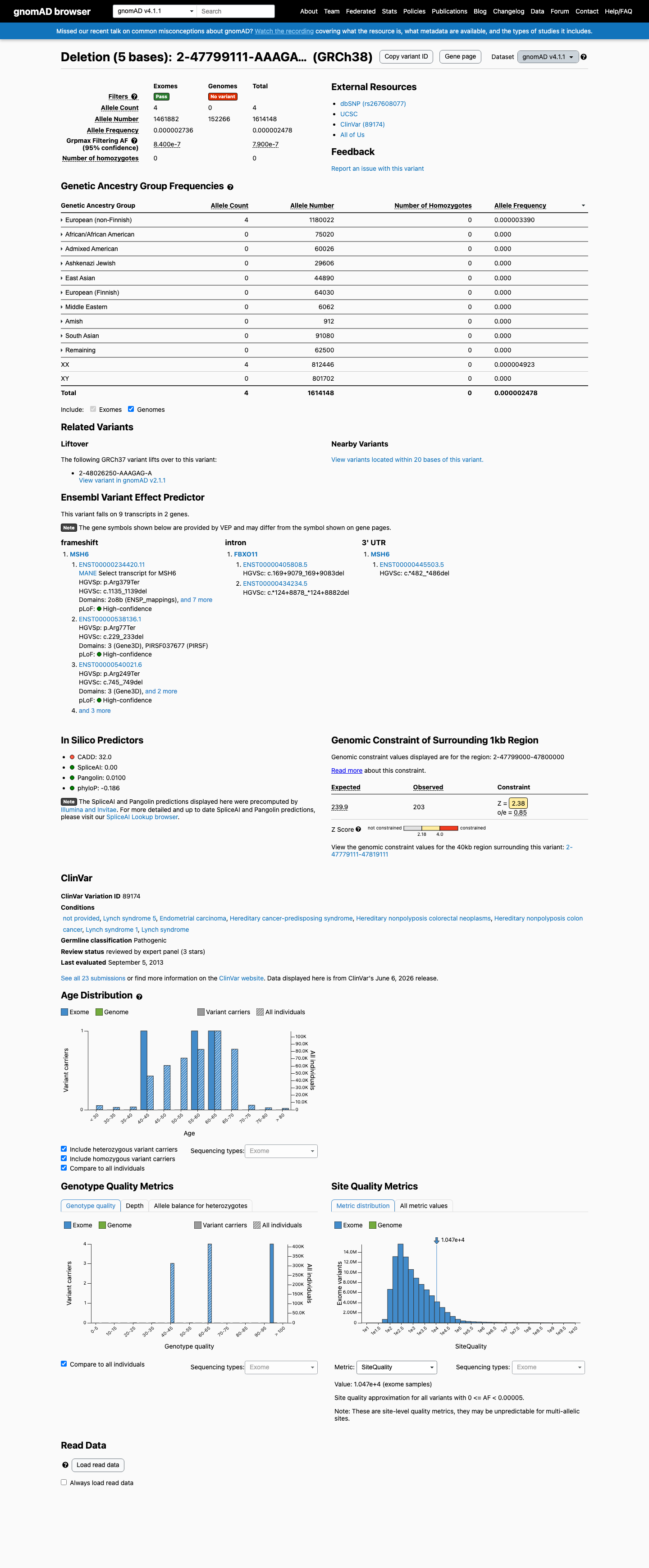
PVS1_Very_Strong: NM_000179.3:c.1135_1139del introduces a premature termination codon at p.Arg379Ter, well within the VCEP boundary of codon 1341. Loss of function is an established disease mechanism for MSH6 in Lynch syndrome.1 PM2_Supporting: This variant is extremely rare in population databases. In gnomAD v4.1, allele frequency is 2.48e-06 (4/1,614,148 alleles, grpmax FAF=7.9e-07), below the VCEP threshold of <0.00002.2 PP4_Moderate: The variant has been observed in a patient with two independent MSH6-deficient tumors (endometrial cancer at age 54 and colorectal cancer at age 58), meeting VCEP criteria for 2 independent tumors with IHC loss consistent with the variant gene. PS2_Supporting: The variant was identified as a likely de novo mosaic variant detectable across all three germ layers in a patient with MSH6-deficient Lynch spectrum tumors, meeting 0.5 de novo points under the VCEP PS2 scoring system. Combining 1 Very Strong (PVS1) + 2 Supporting (PM2 + PS2) satisfies VCEP Rule 4 (1 VS + >=2 Sup → Pathogenic). The additional Moderate criterion (PP4) further supports the pathogenic classification.3
MSH6
Final classification
Pathogenic
MSH6 c.1135_1139del · p.Arg379Ter
MSH6
PVS1_Very_Strong: NM_000179.3:c.1135_1139del introduces a premature termination codon at p.Arg379Ter, well within the VCEP boundary of codon 1341. Loss of function is an established disease mechanism for MSH6 in Lynch syndrome.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MSH6 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule4 (1 Pathogenic.Very Strong + Pathogenic.Supporting >=2) with applied criteria: PVS1 very strong, PS2 supporting, PM2 supporting, PP4 moderate, PP5 supporting; maps to Pathogenic.
Classification rationale
PVS1PS2PM2PP4PP5
Pathogenic
MSH6 c.1135_1139del
PVS1 + PS2 + PM2 + PP4 + PP5
→
Pathogenic
Gene diagram
· NM_000179.3 · variants mapped to exon structure
MSH6
NM_000179.3
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 9 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_000179.3:c.1135_1139del is a 5-bp deletion in exon 4 of MSH6 that introduces a premature termination codon at p.Arg379Ter. Under the InSiGHT/ClinGen MSH6 VCEP v2.0.0, nonsense/frameshift variants introducing a PTC at or before codon 1341 meet PVS1 at Very Strong strength. The PTC at codon 379 lies well within this boundary, and loss of function is an established disease mechanism for MSH6 in Lynch syndrome.
MSH6 loss-of-function is an established mechanism for Lynch syndrome (CSPEC/VCEP confirmed)5-bp deletion creates frameshift introducing PTC at codon 379PTC position 379 ≤ 1341 meets VCEP PVS1 Very Strong threshold
✓
PS2
supporting
Pathogenic
PMID 37318702 reports this variant as a likely de novo mosaic MSH6:c.1135_1139del p.Arg379* variant in a patient with MSH6-deficient endometrial and colorectal cancer. The variant was detected at low mosaic levels in blood (1.64%), saliva (3.49%), and normal colonic tissue (5.34%), indicating early embryonic occurrence consistent with de novo origin. Under the MSH6 VCEP PS2 scoring system, a proband with a de novo variant without confirmed parental testing but clinically likely de novo and with MMR-deficient LS spectrum tumors receives 0.5 points, meeting PS2_Supporting.
PMID 37318702: likely de novo mosaic MSH6:c.1135_1139del in patient with MSH6-deficient EC and CRCVariant detected across all three germ layers confirming early embryonic originMeets VCEP PS2 0.5-point threshold for de novo without parental confirmation
✓
PM2
supporting
Pathogenic
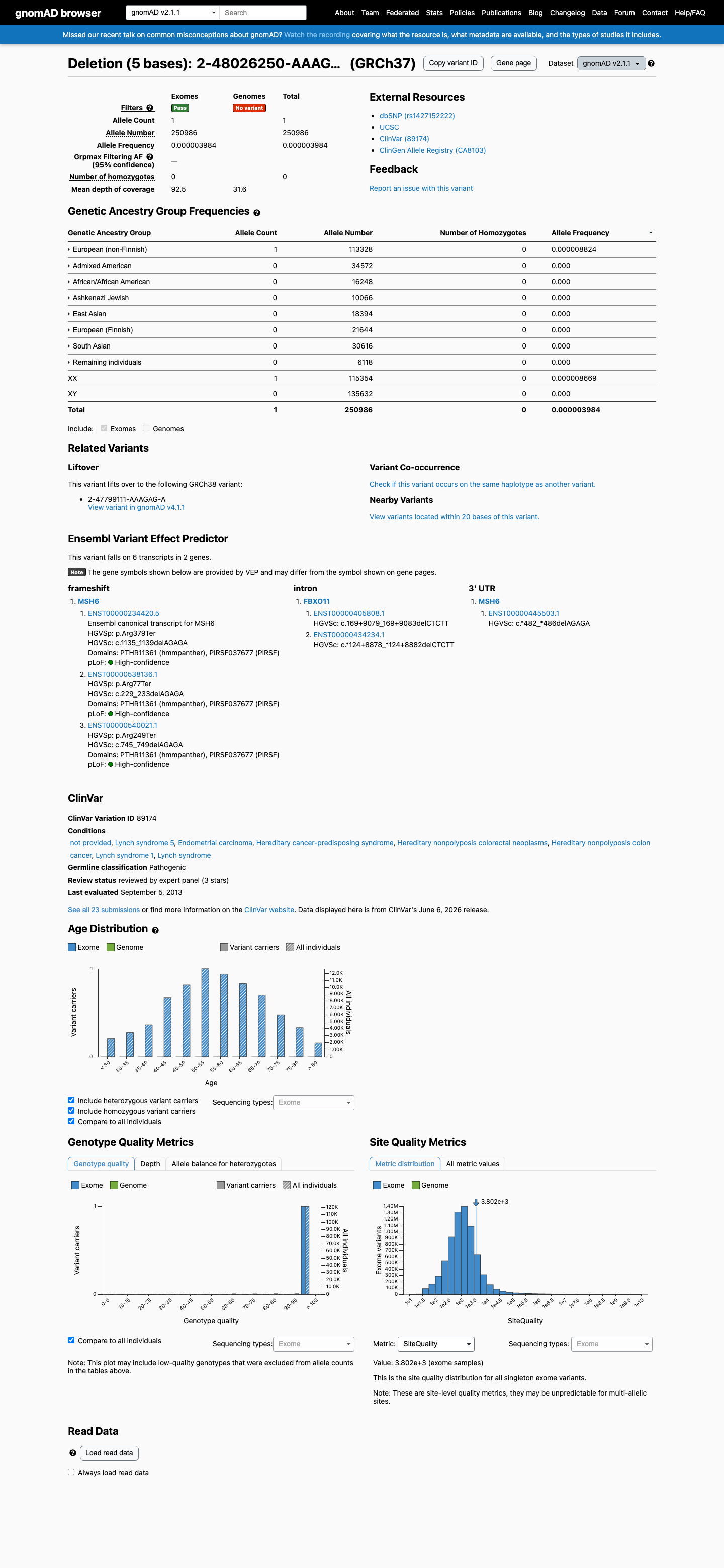
In gnomAD v4.1, this variant has an allele frequency of 2.48e-06 (4/1,614,148 alleles, grpmax FAF=7.9e-07), which is below the VCEP threshold of <0.00002 (<1 in 50,000 alleles). In gnomAD v2.1, it has an AF of 3.98e-06 (1/250,986 alleles). No homozygotes are observed in either dataset. This meets PM2_Supporting per the MSH6 VCEP.
gnomAD v4.1 AF = 2.48e-06 (4/1614148)
✓
PP4
moderate
Pathogenic
The case report in PMID 37318702 describes a patient with two independent MSH6-deficient tumors: endometrial cancer at age 54 and colorectal cancer at age 58. Both tumors showed loss of MSH6 protein expression consistent with the variant location. Under the MSH6 VCEP, 2 independent CRC/Endometrial tumors with loss of MMR protein expression consistent with the variant location meets PP4_Moderate.
PMID 37318702: patient developed MSH6-deficient EC at age 54 and CRC at age 58Both tumors showed MSH6 protein loss consistent with variant locationMeets VCEP PP4_Moderate: 2 independent MSI-H/MSH6-deficient tumors
Assessed · not applied
Pathogenic
PS3
No calibrated functional assay data (functional odds for pathogenicity) is available for this specific variant in the VCEP functional assay SVI documentation.
PP1
No co-segregation data (Bayes Likelihood Ratio from pedigrees) is available for this variant in the evidence collected.
PP3
PP3 under the MSH6 VCEP requires either (a) a missense variant with HCI prior probability >0.68, or (b) a non-canonical splice variant with SpliceAI delta score ≥0.2.
Benign
BA1
The MSH6 VCEP BA1 threshold is gnomAD v4 grpmax FAF ≥ 0.0022 (0.22%).
BS1
The MSH6 VCEP BS1 threshold is gnomAD v4 grpmax FAF ≥ 0.00022 (0.022%) and < 0.0022.
BS2
BS2 under the MSH6 VCEP requires observation of the variant in trans with a known pathogenic MSH6 variant in a patient with CRC after age 45 without CMMRD features, with confirmed phase.
BS3
No calibrated functional assay data demonstrating proficient MMR function (functional odds for pathogenicity ≤0.05 or >0.05 & ≤0.48) is available for this specific variant.
BS4
BS4 under the MSH6 VCEP requires lack of co-segregation with disease in pedigrees with a combined Bayes Likelihood Ratio meeting VCEP thresholds.
BP5
BP5 requires tumors showing MSS and/or no loss of MMR protein expression, or BRAF V600E/MLH1 methylation in MSI-H tumors.
N/A · 13
PS1 · PS4 · PM1 · PM4 · PM5 · PM6 · PP2 · BP1 · BP2 · BP3 · BP4 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.47809e-06; MAF= 0.00025%, 4/1614148 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.38977e-06; MAF= 0.00034%, 4/1180022 alleles, homozygotes = 0); grpmax FAF= 7.9e-07.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.98429e-06; MAF= 0.00040%, 1/250986 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.82394e-06; MAF= 0.00088%, 1/113328 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,614,148
0 hom · FAF 7.9e-05%
0 hom · FAF 7.9e-05%
European (non-Finnish) 4 / 1,180,022 |
0.00034% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 250,986
0 hom
0 hom
European (non-Finnish) 1 / 113,328 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
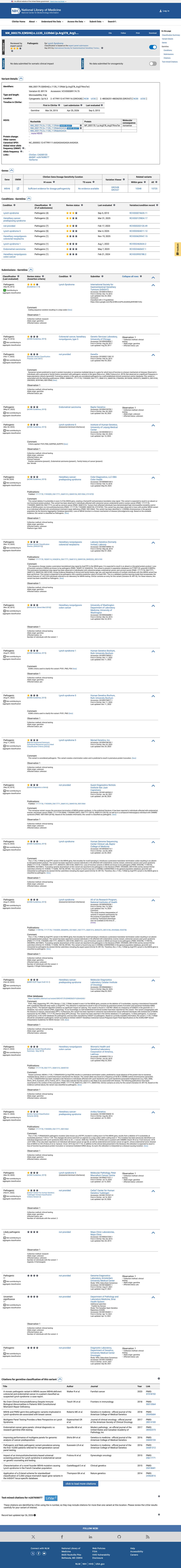
This variant has been reported in ClinVar as Pathogenic (20 clinical laboratories) and as Uncertain significance (1 clinical laboratory) and as Pathogenic by International Society for Gastrointestinal Hereditary Tumours (InSiGHT) (expert panel). (ClinVarID = 89174)
In silico
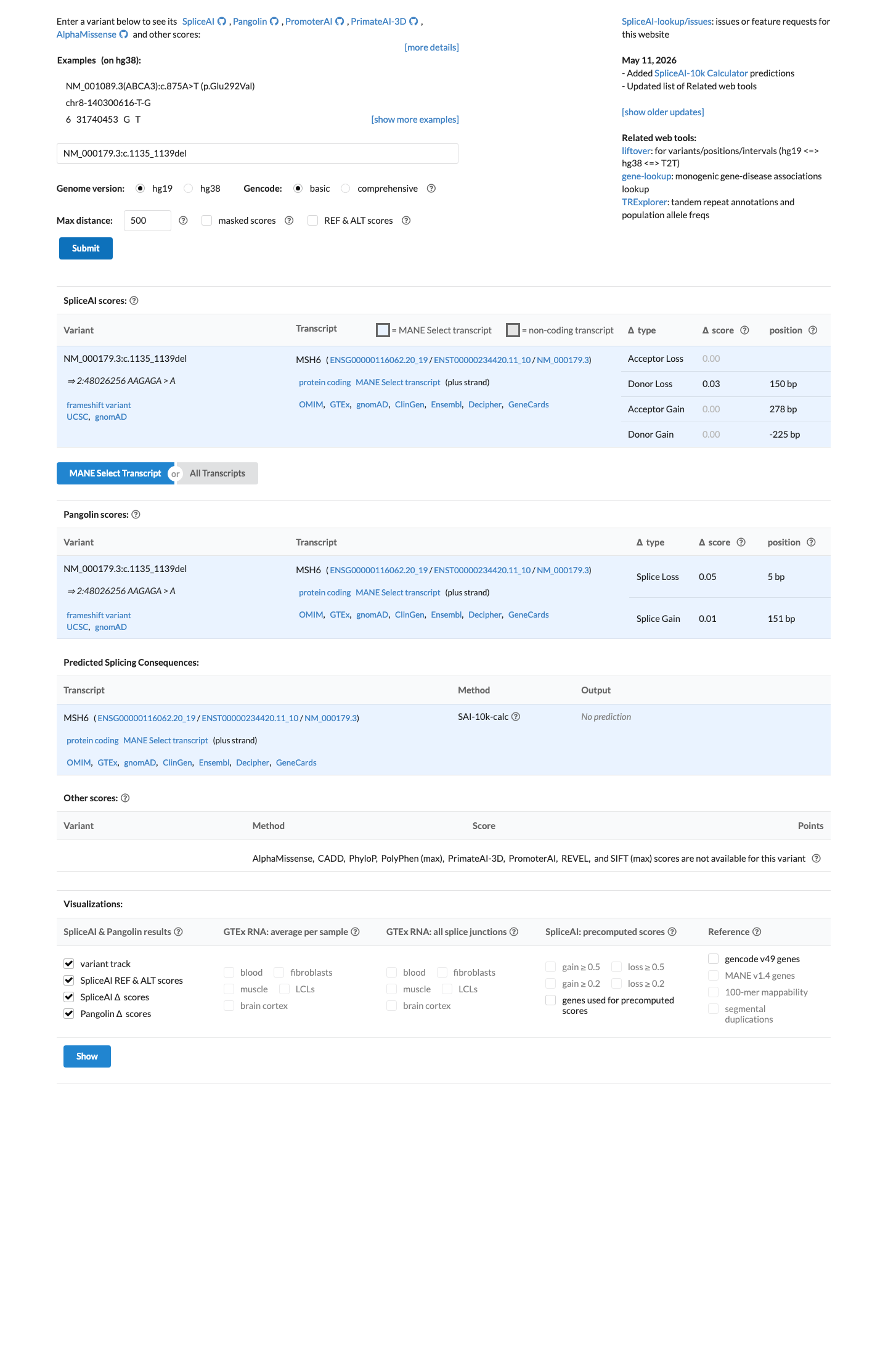
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 8 further PMIDs triaged but not cited — see Sources & References.
PMID 37318702
Found
Structured finding pending for this record — see source link.
Applied to
→PP4 supports · met
→PS2 supports · met
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
24362816 ↗
Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database.
ONCOKB
1651234 ↗
Altering the conserved nucleotide binding motif in the Salmonella typhimurium MutS mismatch repair protein affects both its ATPase and mismatch binding activities.
ONCOKB
24755471 ↗
Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer.
ONCOKB
9111312 ↗
Genetic and biochemical analysis of Msh2p-Msh6p: role of ATP hydrolysis and Msh2p-Msh6p subunit interactions in mismatch base pair recognition.
ONCOKB
9564049 ↗
hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSalpha.
ONCOKB
9822680 ↗
Nucleotide-promoted release of hMutSalpha from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism.
ONCOKB
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR