NM_006164.4:c.100C>G (p.Arg34Gly) is a missense variant in exon 2 of NFE2L2. It is absent from gnomAD v2.1 and v4.1 population databases (PM2_Supporting).1 The variant is located at codon 34 within the Neh2 domain (residues 16–89), the KEAP1-binding degron that is a critical and well-established functional domain. Arg34 is a statistically significant cancer hotspot residue, and other missense alterations at this codon (R34Q, R34P) are recurrently observed in NSCLC and other cancers with no benign variation at this codon (PM1).2 The variant has been reported in COSMIC (COSV67960061, n=42) as a somatic mutation. OncoKB classifies it as Likely Oncogenic (somatic context). These somatic observations do not directly inform germline pathogenicity under ACMG/AMP.3 In silico predictions are equivocal: REVEL score 0.543 (borderline), BayesDel score 0.170 (low), and SpliceAI max delta 0.00. These do not meet the threshold for PP3 or BP4.4 No functional studies, case-control data, de novo observations, segregation data, or authoritative germline classifications are available for this variant. The majority of criteria cannot be assessed. Applying the generic ACMG/AMP 2015 combination rules: PM1 (moderate) + PM2_Supporting (supporting) = 1 moderate + 1 supporting. This is insufficient for a Likely Pathogenic classification (requires 2 moderate, or 1 strong + 1 moderate, or 1 strong + ≥2 supporting). No benign criteria are met. The variant is classified as a Variant of Uncertain Significance (VUS).5
NFE2L2
Final classification
VUS
NFE2L2 c.100C>G · p.Arg34Gly
NFE2L2
NM_006164.4:c.100C>G (p.Arg34Gly) is a missense variant in exon 2 of NFE2L2. It is absent from gnomAD v2.1 and v4.1 population databases (PM2_Supporting).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 supporting; combination = 1 moderate + 1 supporting, which maps to VUS.
Classification rationale
PM1PM2
VUS
NFE2L2 c.100C>G
PM1 + PM2
→
VUS
Gene diagram
· NM_006164.4 · variants mapped to exon structure
NFE2L2
NM_006164.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 19 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
The variant is located at codon 34 (Arg34) within the Neh2 domain (residues 16–89), the KEAP1-binding degron that is a critical and well-established functional domain. Arg34 is a statistically significant cancer hotspot residue (Cancer Hotspots), and other missense alterations at this codon (R34Q, R34P) are recurrently observed in NSCLC and other cancers. The variant is absent from gnomAD population databases, indicating no benign variation at this position.
Located at Arg34 in the Neh2 KEAP1-binding domain (residues 16–89).Cancer Hotspots identifies codon 34 as a statistically significant hotspot.Other missense variants at codon 34 (R34Q/c.101G>A
✓
PM2
supporting
Pathogenic
NM_006164.4:c.100C>G is absent from gnomAD v2.1 and v4.1 population databases (allele frequency <0.1%), meeting the PM2 threshold for a rare variant absent from population controls.
Absent from gnomAD v2.1.Absent from gnomAD v4.1.
Assessed · not applied
Pathogenic
PS1
No other pathogenic missense variant at codon 34 (Arg34) with a different amino acid change was identified for comparison.
PS2
No de novo observation of NM_006164.4:c.100C>G with confirmed maternity/paternity has been reported in the literature or ClinVar.
PS3
No well-established functional assay data are available for NM_006164.4:c.100C>G (p.Arg34Gly) demonstrating a deleterious effect in a germline disease context.
PS4
No case-control studies or enrichment data are available comparing the prevalence of NM_006164.4:c.100C>G in affected individuals versus controls.
PM6
No de novo observation of NM_006164.4:c.100C>G without confirmation of maternity/paternity has been reported.
PP1
No cosegregation data are available for NM_006164.4:c.100C>G in families with NFE2L2-related disease.
PP2
PP2 requires a low rate of benign missense variation in the gene and missense variants as a common disease mechanism.
PP3
In silico predictions do not provide consistent support for a deleterious effect.
PP4
No well-defined germline disease phenotype with high specificity is associated with NFE2L2 variants.
PP5
No reputable source has reported NM_006164.4:c.100C>G as pathogenic.
Benign
BA1
The variant is absent from gnomAD v2.1 and v4.1 population databases.
BS1
The variant is absent from gnomAD v2.1 and v4.1.
BS2
No observation of this variant in healthy adults in the context of a fully penetrant NFE2L2-related disorder has been documented.
BS3
No well-established functional studies demonstrate that NM_006164.4:c.100C>G has no damaging effect on protein function or splicing.
BS4
No data are available on non-segregation of NM_006164.4:c.100C>G with disease in affected families.
BP1
BP1 applies when a missense variant is found in a gene where primarily truncating variants cause disease.
BP4
In silico predictions do not provide consistent evidence for a benign effect.
BP5
No case has been reported in which NM_006164.4:c.100C>G was found in an individual with an alternate molecular basis for disease.
BP6
No reputable source has reported NM_006164.4:c.100C>G as benign.
N/A · 7
PVS1 · PM3 · PM4 · PM5 · BP2 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar but submission details could not be extracted. (ClinVarID = 3258027)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.543. BayesDel score = 0.170416.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
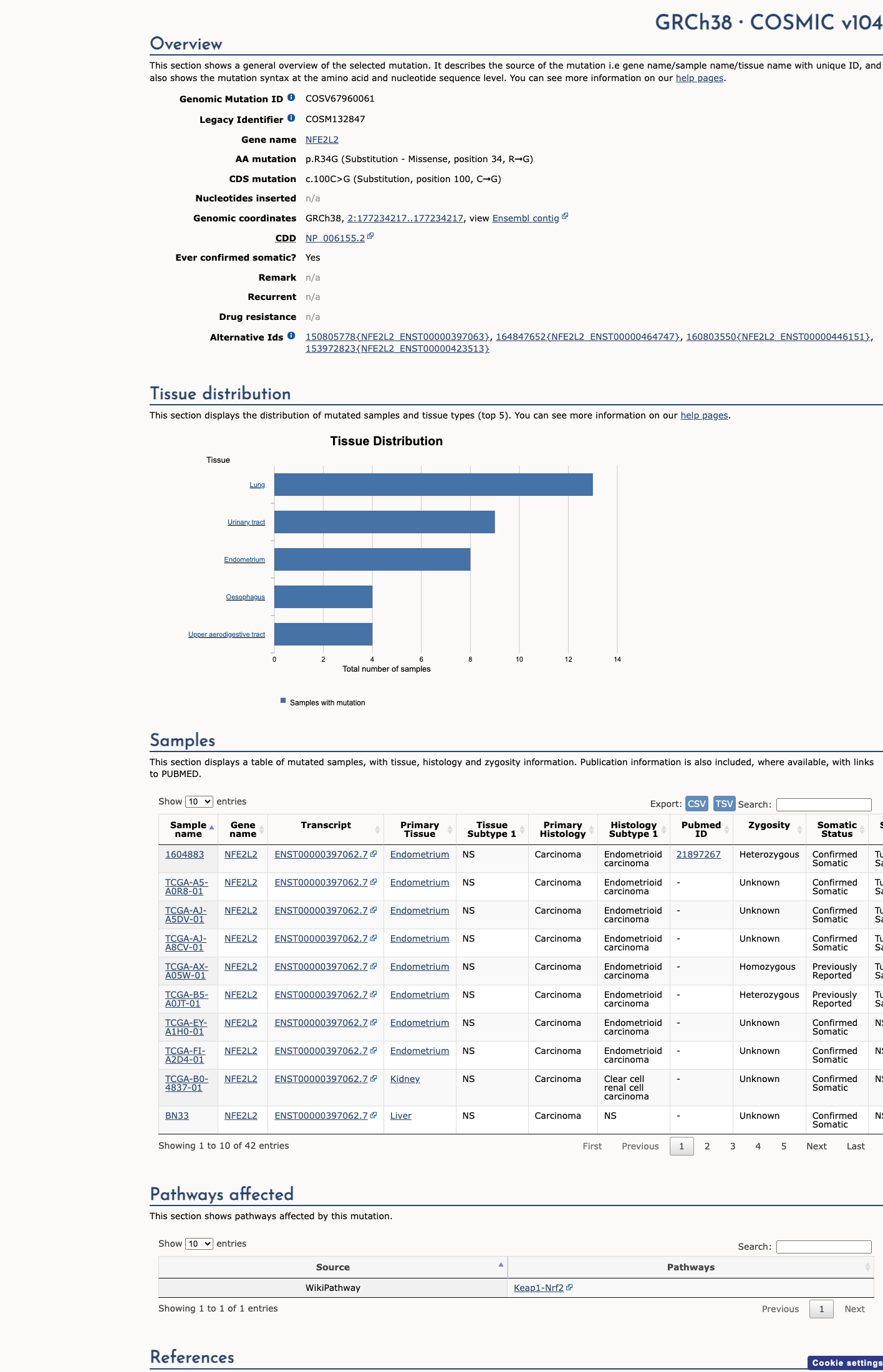
COSMIC
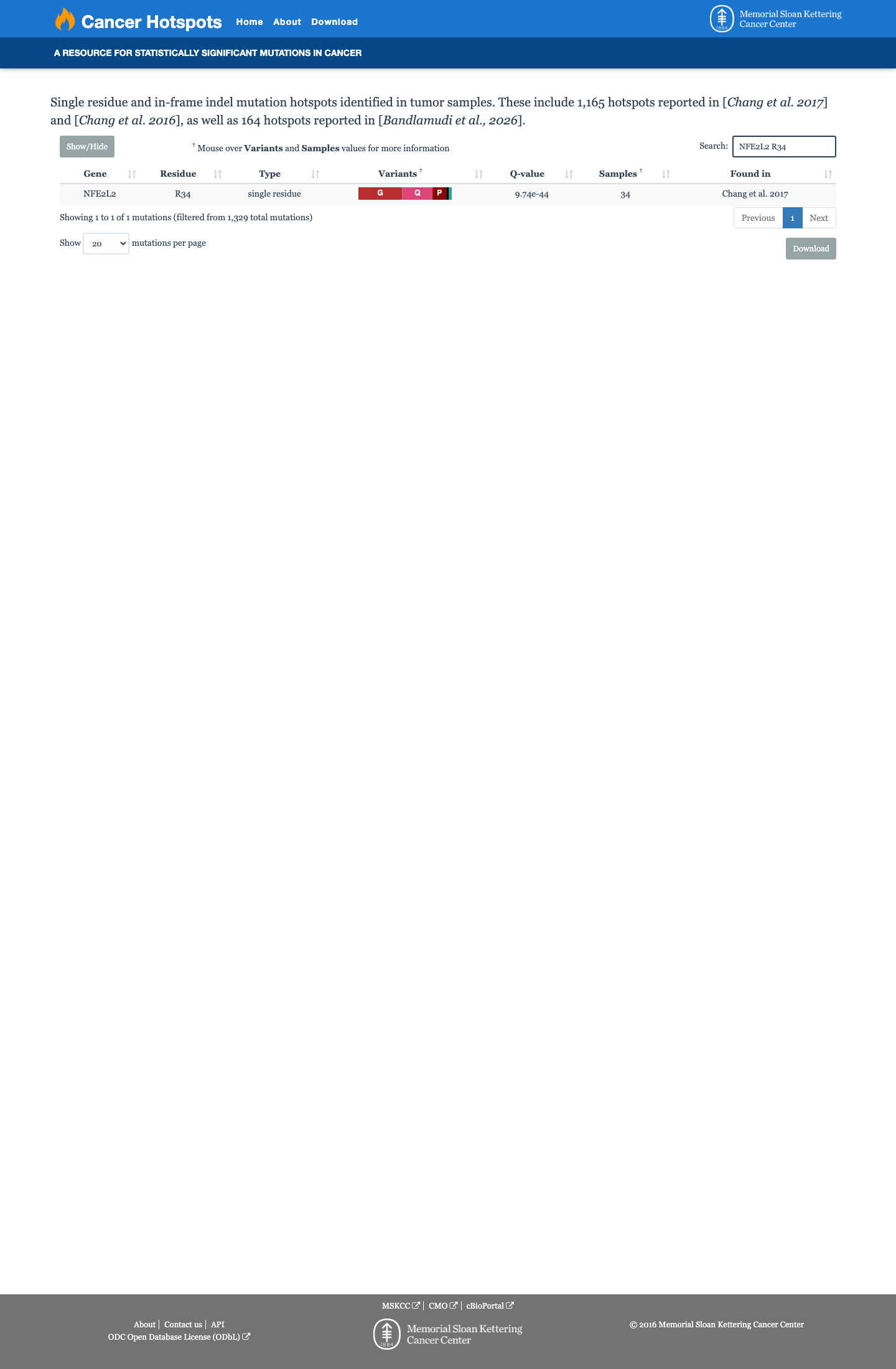
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV67960061, n = 42 times).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Genomic structure and variation of nuclear factor (erythroid-derived 2)-like 2.
Found
Other missense variants at codon 34 (R34Q/c.101G>A R34P/c.101G>C) are reported as somatic mutations in NSCLC and other cancers in PMID:23936606.
Applied to
→PM1 supports · met
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
21897267 ↗
Association of keap1 and nrf2 genetic mutations and polymorphisms with endometrioid endometrial adenocarcinoma survival.
ONCOKB
35101336 ↗
Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC).
CLINVAR
22918138 ↗
Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology.
CLINVAR
34131312 ↗
Chromosomal microarray analysis, including constitutional and neoplastic disease applications, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
23619274 ↗
American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders.
CLINVAR