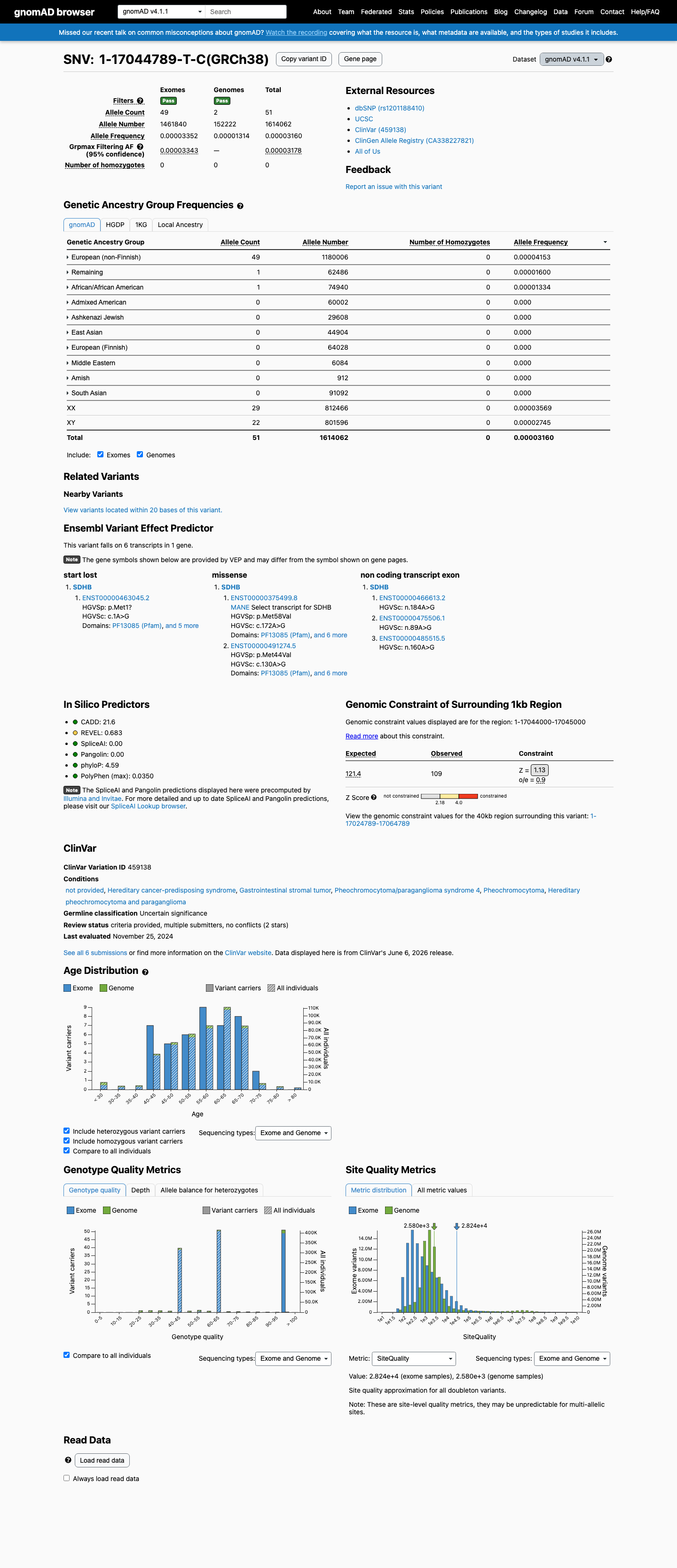
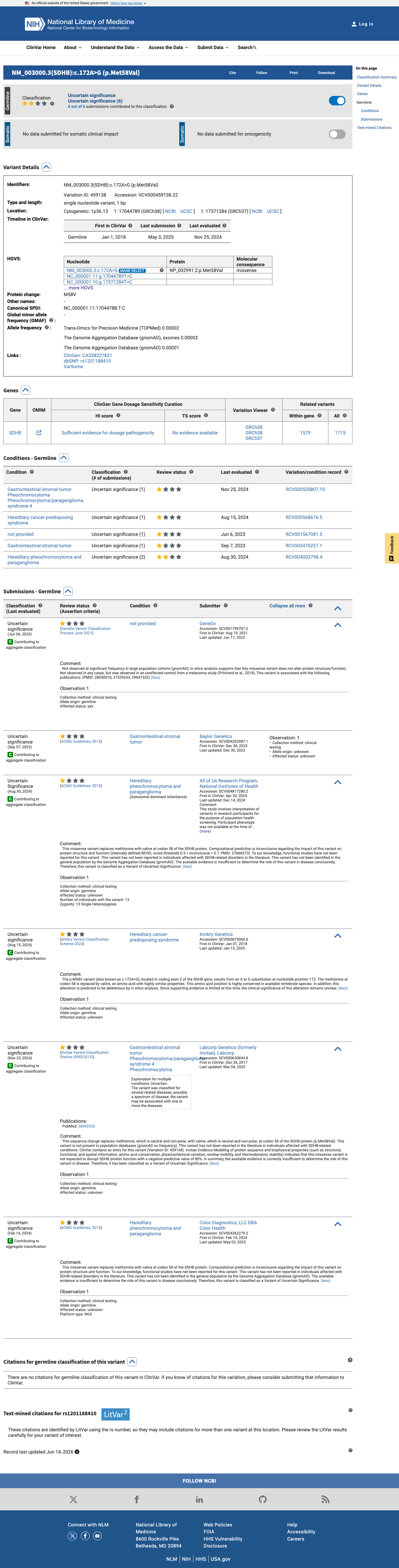
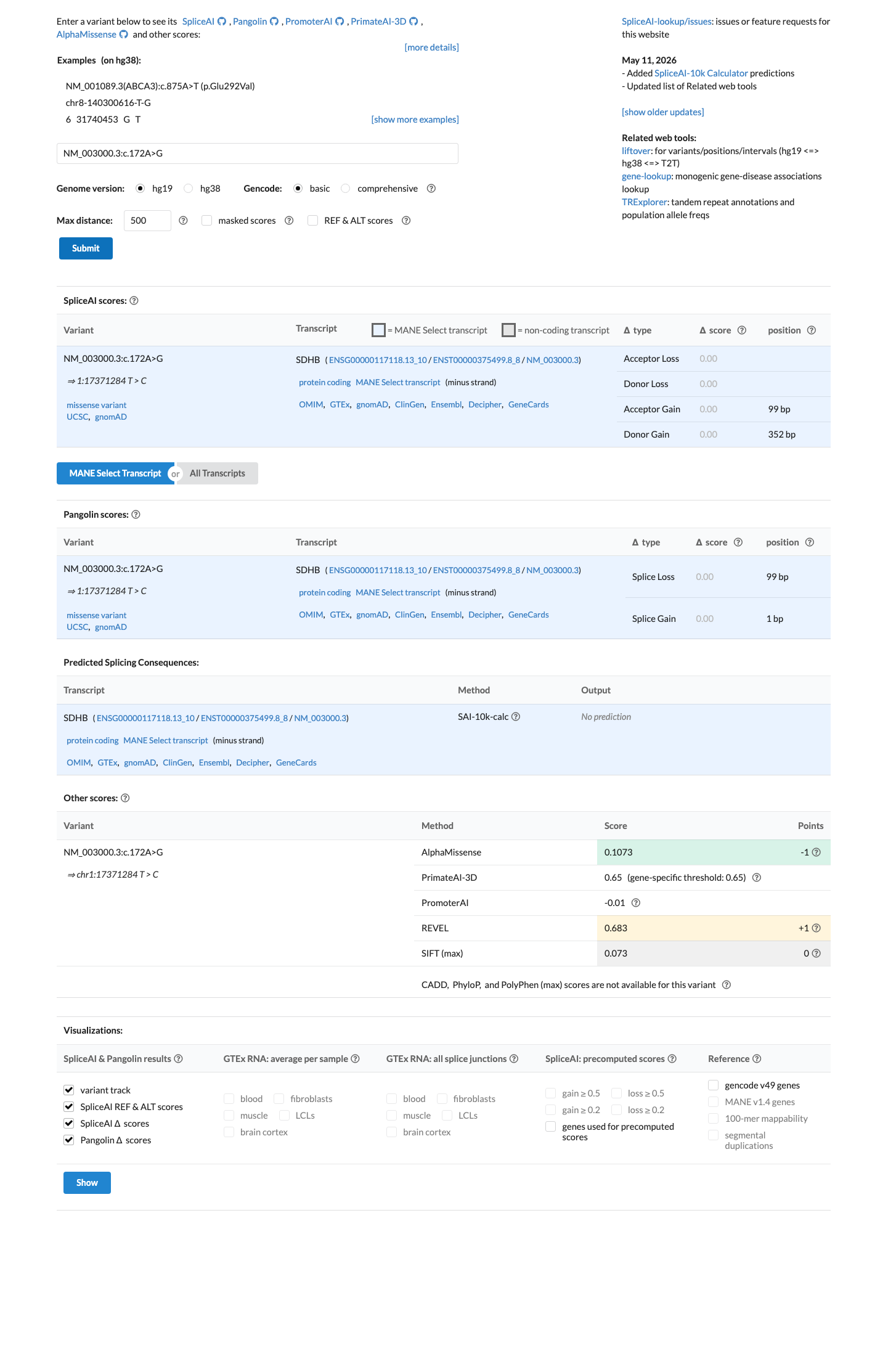
NM_003000.3:c.172A>G (p.Met58Val) is a missense variant in SDHB, a tumor suppressor gene associated with autosomal dominant hereditary pheochromocytoma-paraganglioma.1 This variant is absent from gnomAD v2.1 and v4.1, supporting PM2 at supporting level per generic ACMG/AMP 2015 criteria.2 Six clinical laboratories have submitted this variant to ClinVar, all classifying it as Uncertain Significance (ClinVar variation ID: 459138).3 In silico predictions are indeterminate: SpliceAI predicts no splice impact (max delta 0.00) and BayesDel score is 0.338, which does not reach a pathogenic or benign threshold. REVEL score is unavailable.4 No functional studies, case reports, segregation data, or de novo observations have been identified for this variant in the reviewed literature or curated databases.5 No pathogenic missense variant at the same amino acid residue (Met58) was identified for PM5 comparison.6 The ClinGen Endocrine Tumor Predisposition Expert Panel specifications for SDHB (version 1.0.0) were identified but contained no parsed criterion-level rules; classification adheres to generic ACMG/AMP 2015 framework.7 With only PM2_Supporting met and no additional pathogenic or benign criteria satisfied, the variant remains a Variant of Uncertain Significance per ACMG/AMP 2015 combination rules (PMID:25741868).8
SDHB
Final classification
VUS
SDHB c.172A>G · p.Met58Val
SDHB
NM_003000.3:c.172A>G (p.Met58Val) is a missense variant in SDHB, a tumor suppressor gene associated with autosomal dominant hereditary pheochromocytoma-paraganglioma.
ClinGen Endocrine Tumor Predisposition Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for SDHB Version 1.0.0 v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
SDHB c.172A>G
PM2
→
VUS
1
cspec ↗
4
spliceai ↗bayesdel
5
oncokb ↗
6
pm5_candidates
7
cspec ↗generic_acmg_combination_rules
8
generic_acmg_combination_rules
Gene diagram
· NM_003000.3 · variants mapped to exon structure
SDHB
NM_003000.3
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 20 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_003000.3:c.172A>G is absent from gnomAD v2.1, gnomAD v4.1, and gnomAD-Canada, supporting a low population frequency consistent with a rare pathogenic variant. PM2 at supporting level is applied per generic ACMG/AMP 2015 criteria for variants absent from large population databases.
Absent from gnomAD v2.1 (exomes).Absent from gnomAD v4.1 (exomes).Absent from gnomAD-Canada v1.0.
Assessed · not applied
Pathogenic
PS1
No prior evidence identifies a different nucleotide change at codon 58 resulting in the same amino acid substitution (p.Met58Val) that is classified as pathogenic.
PS2
No de novo observation has been reported for this variant.
PS3
No well-established in vitro or in vivo functional studies have been identified for this specific variant.
PS4
The prevalence of this variant in affected individuals has not been established.
PM1
The variant does not lie within a statistically significant mutational hotspot per Cancer Hotspots analysis, and no evidence establishes residue 58 as a critical or well-established functional domain in SDHB with supporting functional or structural data specific to this position.
PM6
No de novo observation has been reported for this variant.
PP1
No cosegregation data have been reported for this variant.
PP2
PP2 requires quantitative evidence that the gene has a low rate of benign missense variation (e.g., high missense Z-score or low missense constraint metric).
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient-specific phenotype data are available for this variant.
PP5
No reputable source has recently reported this variant as pathogenic.
Benign
BA1
The variant is absent from gnomAD v2.1 and v4.1.
BS1
The variant is absent from gnomAD v2.1 and v4.1.
BS2
No data are available regarding observation of this variant in trans with a pathogenic SDHB variant in a healthy individual.
BS3
No well-established in vitro or in vivo functional studies demonstrate no deleterious effect for this specific variant.
BS4
No segregation data are available for this variant.
BP2
No observation of this variant in trans with a pathogenic SDHB variant in a healthy individual has been reported.
BP4
Multiple lines of computational evidence do not support a benign effect.
BP5
No evidence demonstrates that this variant was found in a case with an alternate molecular basis for disease.
BP6
No reputable source has reported this variant as benign.
N/A · 7
PVS1 · PM3 · PM4 · PM5 · BP1 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (5 clinical laboratories) and as Uncertain Significance (1 clinical laboratory). (ClinVarID = 459138)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.338173.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. SDHB, a subunit of succinate dehydrogenase, is infrequently altered in cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 12 PMIDs not cited in assessment
20816580 ↗
Endocrine cancer predisposition syndromes: hereditary paraganglioma, multiple endocrine neoplasia type 1, multiple endocrine neoplasia type 2, and hereditary thyroid cancer.
CLINVAR
23852704 ↗
Tumor markers in colorectal cancer, gastric cancer and gastrointestinal stromal cancers: European group on tumor markers 2014 guidelines update.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
20065170 ↗
American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility.
CLINVAR
20664475 ↗
The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
22685257 ↗
The UK NEQAS for Molecular Genetics scheme for gastrointestinal stromal tumour: findings and recommendations following four rounds of circulation.
CLINVAR
24893135 ↗
Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
33939658 ↗
The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma.
CLINVAR