NM_000548.5:c.729C>G (p.Leu243=) is a synonymous variant in exon 8 of TSC2 with an overall allele frequency of 0.316-0.355% in gnomAD, exceeding the BS1 threshold of >0.3%.1 This variant has been observed in 10 homozygous individuals in gnomAD v4.1 among 1,612,278 alleles, constituting strong evidence for benignity in a dominant disorder (BS2).2 SpliceAI predicts no splice impact (max delta = 0.01), and the variant is synonymous with no predicted effect on protein sequence, supporting multiple benign computational evidence lines (BP4, BP7).3 ClinVar classifies this variant as Benign/Likely benign across 21 clinical laboratory submissions (15 Benign, 6 Likely benign), with criteria provided and no conflicting pathogenic interpretations (BP6).4 No pathogenic criteria are met. PVS1, PS1, and PM5 are not applicable to this synonymous variant. PS2, PS3, PS4, PS5, PM1, PM2, PM6, PP3, and PP5 are not met. PP1, PP4, BS4, BP2, and BP5 are not assessed due to absence of data. BA1 is not met (overall AF <1%). BP1 and PP2 are not applicable to synonymous variants.5 Applying generic ACMG/AMP 2015 final combination rules: two strong benign criteria (BS1, BS2) alone satisfy the classification threshold for Benign. Additionally, three supporting benign criteria (BP4, BP6, BP7) reinforce the benign classification.6
TSC2
Final classification
Benign
TSC2 c.729C>G · p.Leu243=
TSC2
NM_000548.5:c.729C>G (p.Leu243=) is a synonymous variant in exon 8 of TSC2 with an overall allele frequency of 0.316-0.355% in gnomAD, exceeding the BS1 threshold of >0.3%.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; combination = 2 strong benign + 3 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4BP6BP7
Benign
TSC2 c.729C>G
BS1 + BS2 + BP4 + BP6 + BP7
→
Benign
5
pvs1_generic_framework ↗generic_acmg_combination_rules
6
generic_acmg_combination_rules
Gene diagram
· NM_000548.5 · variants mapped to exon structure
TSC2
NM_000548.5
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 15 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The allele frequency of NM_000548.5:c.729C>G exceeds the BS1 threshold of >0.3% in the general population. gnomAD v2.1: 0.355% (1000/281,464 alleles, 0 homozygotes), gnomAD v4.1: 0.316% (5,099/1,612,278 alleles, 10 homozygotes). This frequency is substantially greater than expected for TSC2-associated tuberous sclerosis complex (incidence ~1:6,000-10,000).
gnomAD v2.1: AF=0.355% (>0.3% BS1 threshold)1000/281464 allelesv4.1: AF=0.316%
✓
BS2
strong
Benign
NM_000548.5:c.729C>G has been observed in 10 homozygous individuals in gnomAD v4.1 (total AF=0.316%, 5,099 heterozygous alleles). Tuberous sclerosis complex is a dominant disorder with high penetrance. Observation of homozygotes and thousands of heterozygotes in a population database of generally healthy individuals constitutes strong evidence for a benign role.
gnomAD v4.1: 10 homozygotes observed5099 total alternate alleles. Dominant disorder observation in healthy population.
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no impact on gene product. SpliceAI predicts no significant splice alteration (max delta score = 0.01). NM_000548.5:c.729C>G is a synonymous variant (p.Leu243=) at a nucleotide position where in silico splicing predictions support a null effect.
SpliceAI max delta = 0.01 (no predicted splice impact)synonymous substitution with no predicted effect on protein sequence or splicing.
✓
BP6
supporting
Benign
Multiple reputable clinical laboratories consistently classify NM_000548.5:c.729C>G as Benign or Likely benign in ClinVar (Variation ID 49382). Fifteen clinical laboratories classify as Benign and six as Likely benign, with criteria provided. No submitting laboratory has asserted pathogenicity.
ClinVar Variation ID 49382: Benign by 15 clinical labsLikely benign by 6 clinical labsmultiple submitters with criteria provided
✓
BP7
supporting
Benign
NM_000548.5:c.729C>G is a synonymous variant (p.Leu243=) for which splicing prediction algorithms predict no impact on splicing. SpliceAI max delta score is 0.01, well below the 0.10 threshold for predicted splice alteration. The nucleotide is not evolutionarily constrained to a degree suggesting functional importance.
Synonymous variant p.(Leu243=)SpliceAI max delta = 0.01 (no predicted splice impact)nucleotide position not at a conserved splice-relevant site.
Assessed · not applied
Pathogenic
PS2
No de novo occurrence with confirmed maternity and paternity has been reported for NM_000548.5:c.729C>G.
PS3
No well-established in vitro or in vivo functional studies have been identified that demonstrate a damaging effect of NM_000548.5:c.729C>G on TSC2 function or splicing.
PS4
No case-control studies demonstrating statistically significant enrichment of NM_000548.5:c.729C>G in affected individuals compared to controls have been identified.
PM1
NM_000548.5:c.729C>G is not located in a recognized mutational hotspot or well-established critical functional domain of TSC2 without benign variation.
PM2
NM_000548.5:c.729C>G is present in gnomAD at allele frequencies well above the 0.1% PM2 threshold.
PM6
No de novo occurrence of NM_000548.5:c.729C>G has been reported without confirmation of paternity/maternity.
PP1
No cosegregation data are available for NM_000548.5:c.729C>G.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No specific patient phenotype data are available for independent review.
PP5
PP5 requires a reputable source to report the variant as pathogenic.
Benign
BA1
The overall allele frequency of NM_000548.5:c.729C>G in gnomAD is below the BA1 threshold of >1%.
BS3
No well-established in vitro or in vivo functional studies demonstrating no deleterious effect of NM_000548.5:c.729C>G have been identified.
BS4
No cosegregation data in affected families are available for NM_000548.5:c.729C>G.
BP2
No data are available regarding observation of NM_000548.5:c.729C>G in trans with a known pathogenic TSC2 variant.
BP5
No case has been identified where NM_000548.5:c.729C>G was found in an individual with an alternate molecular basis for disease.
N/A · 5
PVS1 · PS1 · PM5 · PP2 · BP1
Research & evidence
Population frequency · supports benign
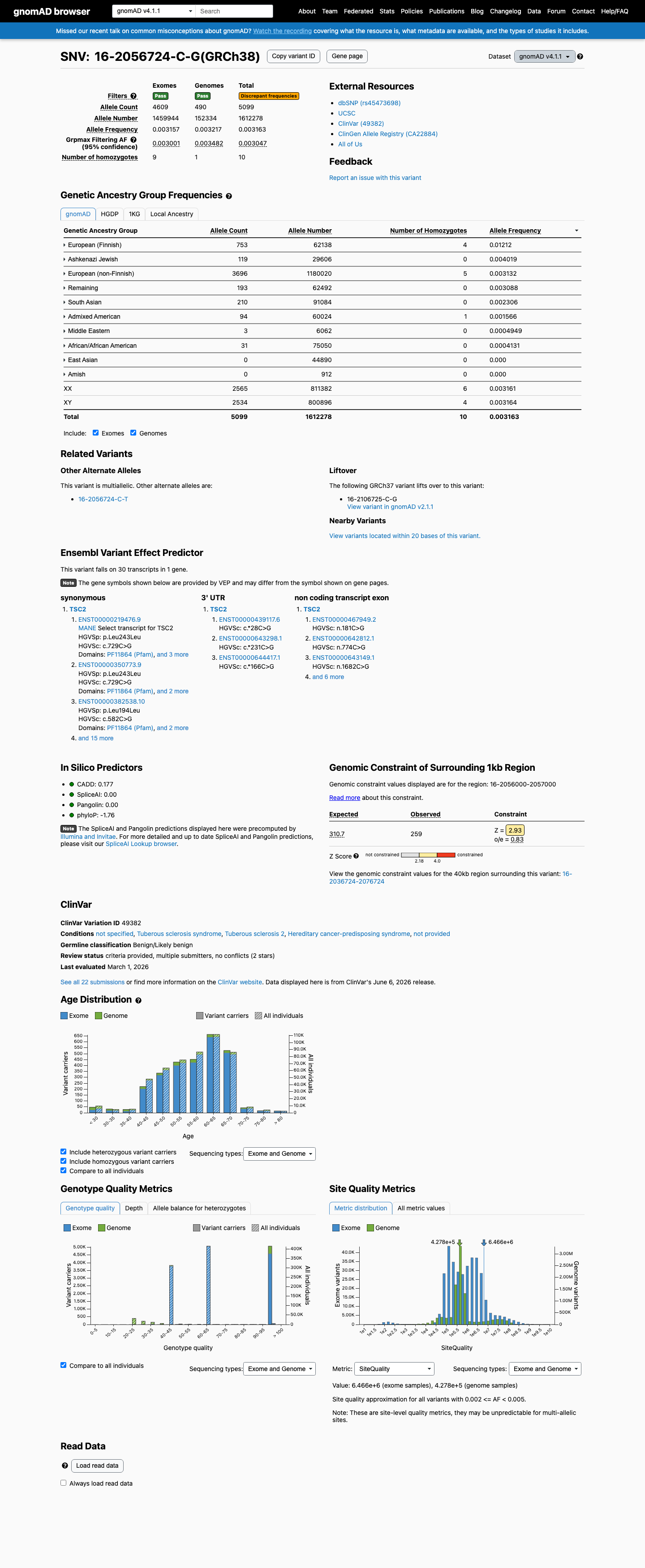
gnomAD v4.1
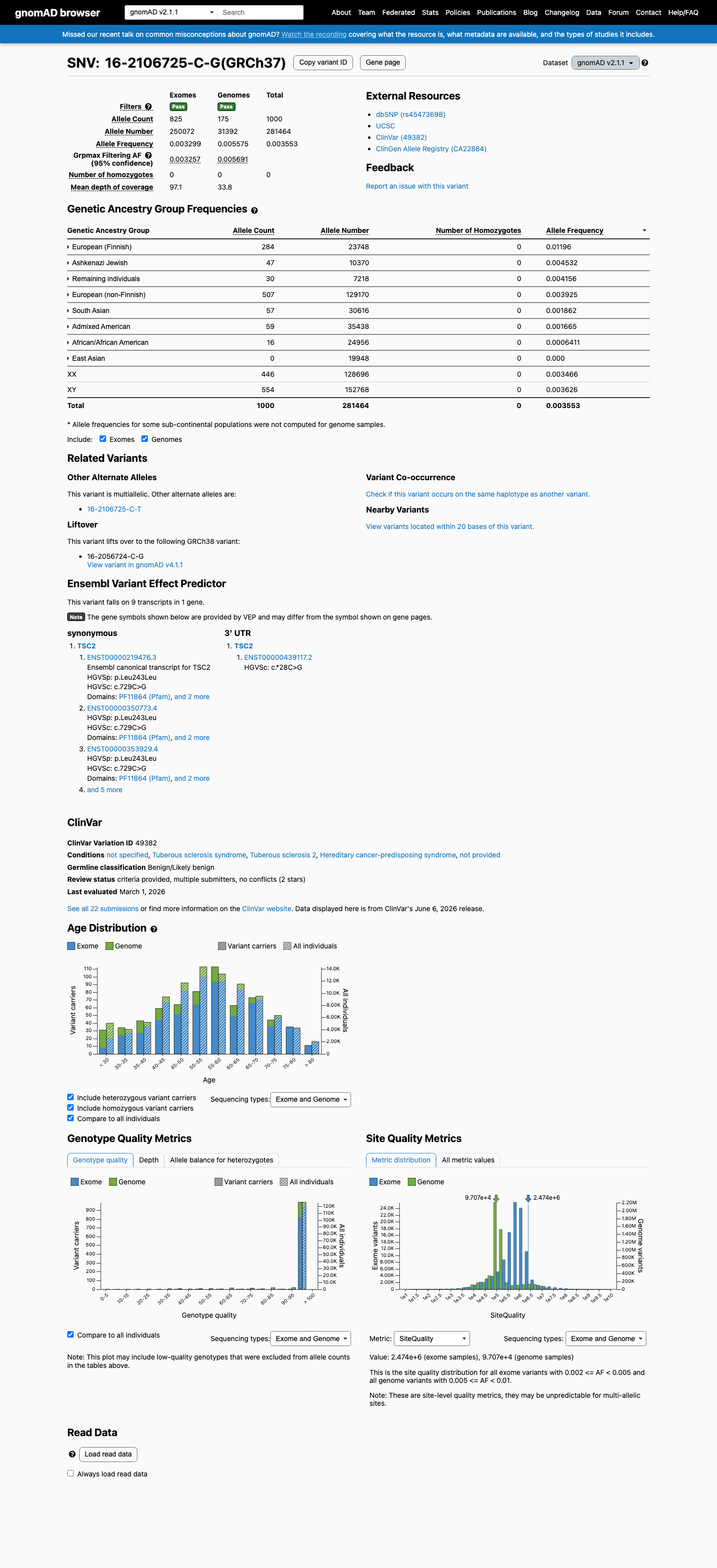
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00316261; MAF= 0.31626%, 5099/1612278 alleles, homozygotes = 10) and has highest observed frequency in the European (Finnish) population (AF= 0.0121182; MAF= 1.21182%, 753/62138 alleles, homozygotes = 4); grpmax FAF= 0.00304706.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00355285; MAF= 0.35529%, 1000/281464 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 0.0119589; MAF= 1.19589%, 284/23748 alleles, homozygotes = 0); grpmax FAF= 0.0056912.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.32%
· 5099 / 1,612,278
10 hom · FAF 0.3%
10 hom · FAF 0.3%
European (Finnish) 753 / 62,138 |
1.2% 4 hom |
Ashkenazi Jewish 119 / 29,606 |
0.4% |
European (non-Finnish) 3696 / 1,180,020 |
0.31% 5 hom |
Remaining individuals 193 / 62,492 |
0.31% |
South Asian 210 / 91,084 |
0.23% |
Admixed American 94 / 60,024 |
0.16% 1 hom |
Middle Eastern 3 / 6,062 |
0.049% |
African/African American 31 / 75,050 |
0.041% |
+ 2 not observed (Amish, East Asian)
gnomAD v2.1
0.36%
· 1000 / 281,464
0 hom · FAF 0.57%
0 hom · FAF 0.57%
European (Finnish) 284 / 23,748 |
1.2% |
Ashkenazi Jewish 47 / 10,370 |
0.45% |
Remaining individuals 30 / 7,218 |
0.42% |
European (non-Finnish) 507 / 129,170 |
0.39% |
South Asian 57 / 30,616 |
0.19% |
Admixed American 59 / 35,438 |
0.17% |
African/African American 16 / 24,956 |
0.064% |
+ 1 not observed (East Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
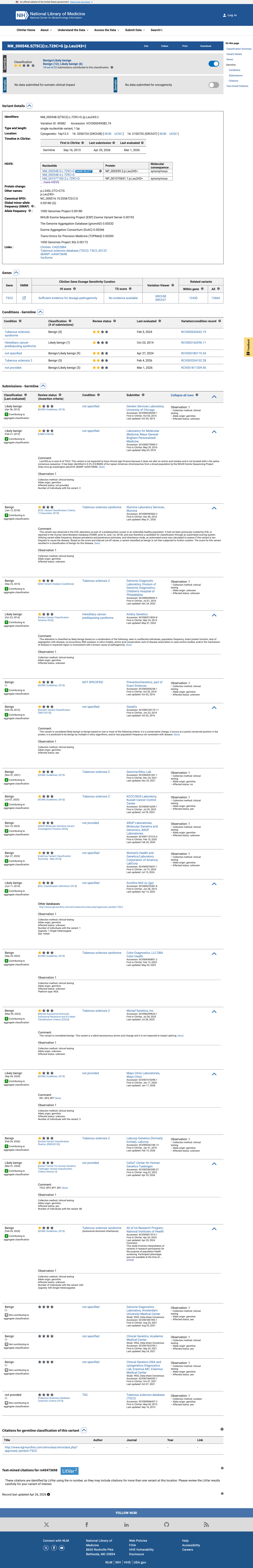
ClinVar
This variant has been reported in ClinVar as Benign (15 clinical laboratories) and as Likely benign (6 clinical laboratories). (ClinVarID = 49382)
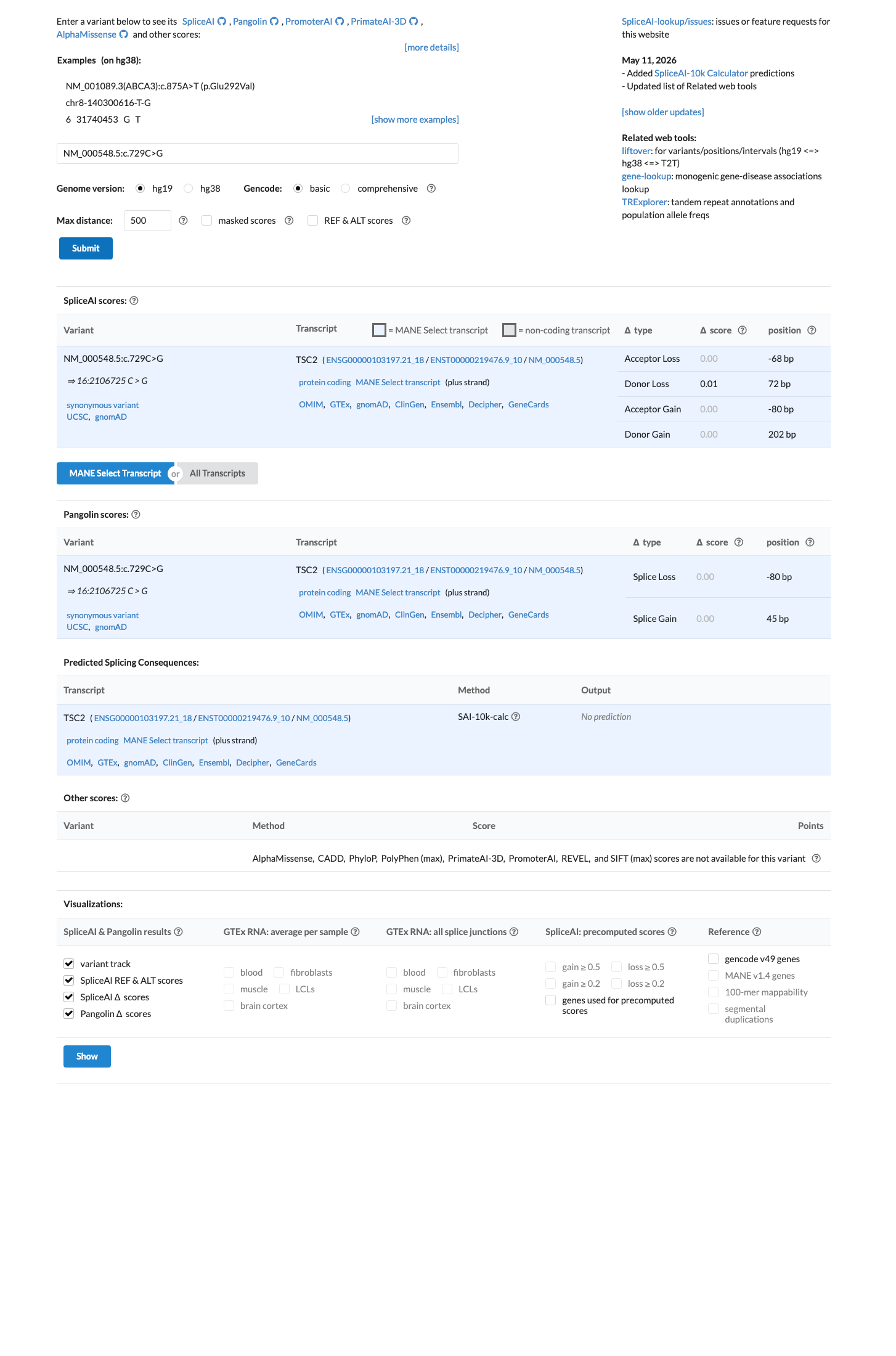
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV104587071, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
15798777 ↗
Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex.
CLINVAR
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
9829910 ↗
Exon scanning of the entire TSC2 gene for germline mutations in 40 unrelated patients with tuberous sclerosis.
CLINVAR
23519317 ↗
Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR