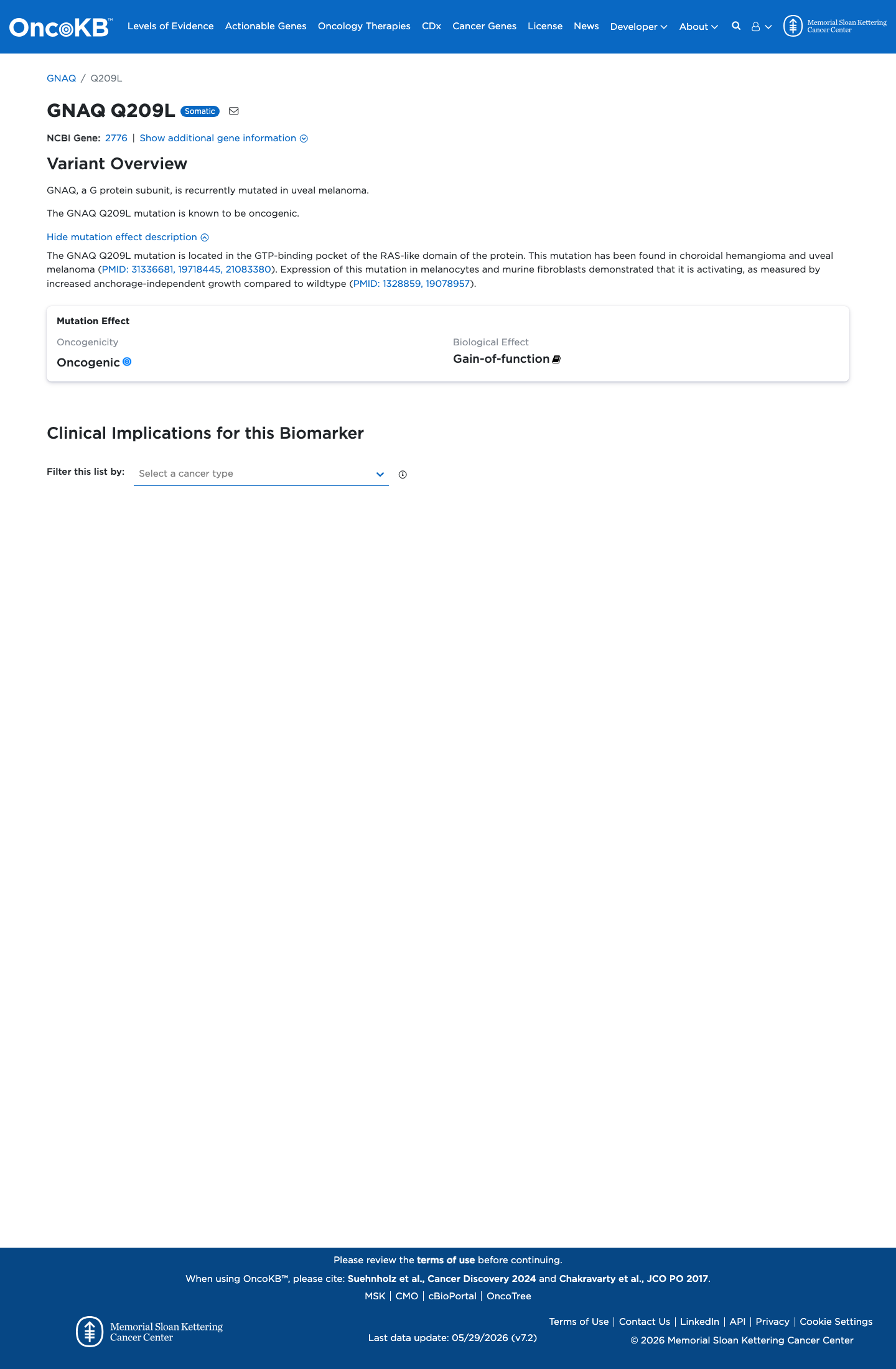
NM_002072.5:c.626A>T (p.Gln209Leu) is a well-characterized gain-of-function missense variant in GNAQ. Multiple independent functional studies demonstrate GTPase deficiency, constitutive activation of downstream MAPK/ERK signaling, and malignant transformation (PS3_strong).1 The variant is located at codon 209, a statistically significant mutational hotspot in the GTPase domain of GNAQ where multiple pathogenic missense substitutions (Q209L, Q209P, Q209H, Q209R) cluster (PM1).2 The variant is absent from all large population databases including gnomAD v2.1, v4.1, and gnomAD-Canada (PM2).3 Other pathogenic missense changes at the same residue (Gln209Pro, Gln209Arg, Gln209His) have been established as pathogenic (PM5).4 GNAQ has a low rate of benign missense variation and missense variants at residues 183 and 209 are the established gain-of-function disease mechanism (PP2).5 Multiple in silico tools predict a deleterious effect: REVEL score 0.936, BayesDel consistent with damaging prediction (PP3).6 ClinVar classifies this variant as Pathogenic (variation ID 375955, one clinical laboratory with criteria provided) (PP5).7 Applying the ACMG/AMP 2015 generic combination rules: 1 Strong (PS3) + 3 Moderate (PM1, PM2, PM5) + 3 Supporting (PP2, PP3, PP5) satisfies the threshold for Pathogenic.8
GNAQ
Final classification
Pathogenic
GNAQ c.626A>T · p.Gln209Leu
GNAQ
NM_002072.5:c.626A>T (p.Gln209Leu) is a well-characterized gain-of-function missense variant in GNAQ. Multiple independent functional studies demonstrate GTPase deficiency, constitutive activation of downstream MAPK/ERK signaling, and malignant transformation (PS3_strong).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PM1 moderate, PM2 moderate, PM5 moderate, PP2 supporting, PP3 supporting, PP5 supporting; combination = 1 strong + 3 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PS3PM1PM2PM5PP2PP3PP5
Pathogenic
GNAQ c.626A>T
PS3 + PM1 + PM2 + PM5 + PP2 + PP3 + PP5
→
Pathogenic
6
revelbayesdel
8
generic_acmg_combination_rules
Gene diagram
· NM_002072.5 · variants mapped to exon structure
GNAQ
NM_002072.5
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 15 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Multiple well-established in vitro and in vivo functional studies from independent groups demonstrate that the Gln209Leu substitution in GNAQ causes GTPase deficiency, constitutive activation of downstream signaling, and malignant transformation. This gain-of-function effect is a well-replicated finding constituting strong functional evidence of a damaging effect.
PMID:1328859: Demonstrated that Q209L Gαq is GTPase-deficient and induces malignant transformation in NIH 3T3 cells.PMID:19078957: Identified Q209L as a frequent activating somatic mutation in uveal melanoma (~50%).PMID:19718445: Confirmed GNAQ codon 209 mutations cause constitutive activation across multiple tumor types.
✓
PM1
moderate
Pathogenic
Codon 209 is a well-established mutational hotspot in GNAQ, located in the GTPase domain. Cancer Hotspots confirms statistically significant clustering at this residue. Multiple publications identify codon 209 as one of two major GNAQ hotspot loci (along with codon 183).
Cancer Hotspots identifies codon 209 as a statistically significant hotspot.PMID:19078957 and PMID:19718445 confirm codon 209 as a recurrently mutated residue in human neoplasia.Multiple pathogenic missense variants cluster at this residue (Q209L
✓
PM2
moderate
Pathogenic
NM_002072.5:c.626A>T is absent from all large population databases, including gnomAD v2.1 (exomes), gnomAD v4.1 (exomes/genomes), and gnomAD-Canada v1.0. Allele frequency is well below the 0.1% threshold for PM2.
Absent from gnomAD v2.1 (AF=0).Absent from gnomAD v4.1 (AF=0).Absent from gnomAD-Canada v1.0.
✓
PM5
moderate
Pathogenic
Other pathogenic missense variants have been established at the same amino acid residue (Gln209): p.Gln209Pro (Q209P), p.Gln209Arg (Q209R), and p.Gln209His (Q209H). PMID:19718445 reports multiple substitutions at codon 209 as constitutively activating. PMID:31336681 specifically identifies Q209R as pathogenic.
PMID:19718445: Multiple mutations at codon 209 cause constitutive GNAQ activation.PMID:31336681: Q209R identified as highly specific pathogenic variant in circumscribed choroidal hemangioma.Q209P and Q209H are known pathogenic alternate substitutions at the same residue.
✓
PP2
supporting
Pathogenic
GNAQ has a low rate of benign missense variation and missense variants at key residues (codons 183 and 209) are the established mechanism of disease. The complete absence of this variant from gnomAD is consistent with high missense constraint.
GNAQ missense variants at residues 183 and 209 are the known gain-of-function disease mechanism.The variant is absent from large population databasesconsistent with purifying selection against missense changes at functional residues.
✓
PP3
supporting
Pathogenic
REVEL score of 0.936 strongly predicts a deleterious effect. BayesDel (noAF) score is also consistent with a damaging prediction. SpliceAI predicts no splicing impact (max delta 0.03), but this does not contradict the pathogenicity of the missense change.
REVEL: 0.936 (strongly deleterious).BayesDel (noAF): 0.363 (consistent with deleterious).SpliceAI: max delta 0.03 (no splicing impact).
✓
PP5
supporting
Pathogenic
ClinVar reports NM_002072.5:c.626A>T (p.Gln209Leu) as Pathogenic (variation ID 375955). One clinical laboratory (Clinical Genomics Laboratory, Washington University in St. Louis) submitted this classification with criteria provided.
ClinVar variation ID 375955: Pathogeniccriteria providedsingle submitter.
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change producing the same Gln209Leu substitution with established pathogenicity has been identified.
PS2
No confirmed de novo germline occurrence of NM_002072.5:c.626A>T has been reported.
PS4
The variant is absent from population databases (gnomAD) and highly prevalent in somatic tumor cohorts (COSMIC n=346), but no formal germline case-control study is available.
PM6
No de novo occurrence (germline or mosaic) of NM_002072.5:c.626A>T with unconfirmed parentage has been reported.
PP1
No co-segregation data are available.
PP4
No patient phenotype or clinical history was provided with this case.
Benign
BA1
NM_002072.5:c.626A>T is absent from gnomAD v2.1 and v4.1.
BS1
NM_002072.5:c.626A>T is absent from gnomAD.
BS2
No observation of NM_002072.5:c.626A>T in healthy adult individuals has been reported.
BS3
Well-established functional studies universally demonstrate a gain-of-function, damaging effect for the Gln209Leu substitution (GTPase deficiency, constitutive signaling, transformation).
BP1
GNAQ disease is driven by gain-of-function missense variants, not truncating loss-of-function variants.
BP2
NM_002072.5:c.626A>T has not been observed in trans with a pathogenic variant in a dominant disorder or in cis with a known pathogenic GNAQ variant.
BP4
REVEL score of 0.936 strongly predicts a deleterious effect, directly contradicting the BP4 requirement of multiple lines of computational evidence suggesting no impact.
BP5
No alternate molecular basis for disease has been identified in any case carrying NM_002072.5:c.626A>T.
BP6
ClinVar classifies NM_002072.5:c.626A>T as Pathogenic, not benign.
N/A · 6
PVS1 · PM3 · PM4 · BS4 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Pathogenic (1 clinical laboratory). (ClinVarID = 375955)
In silico
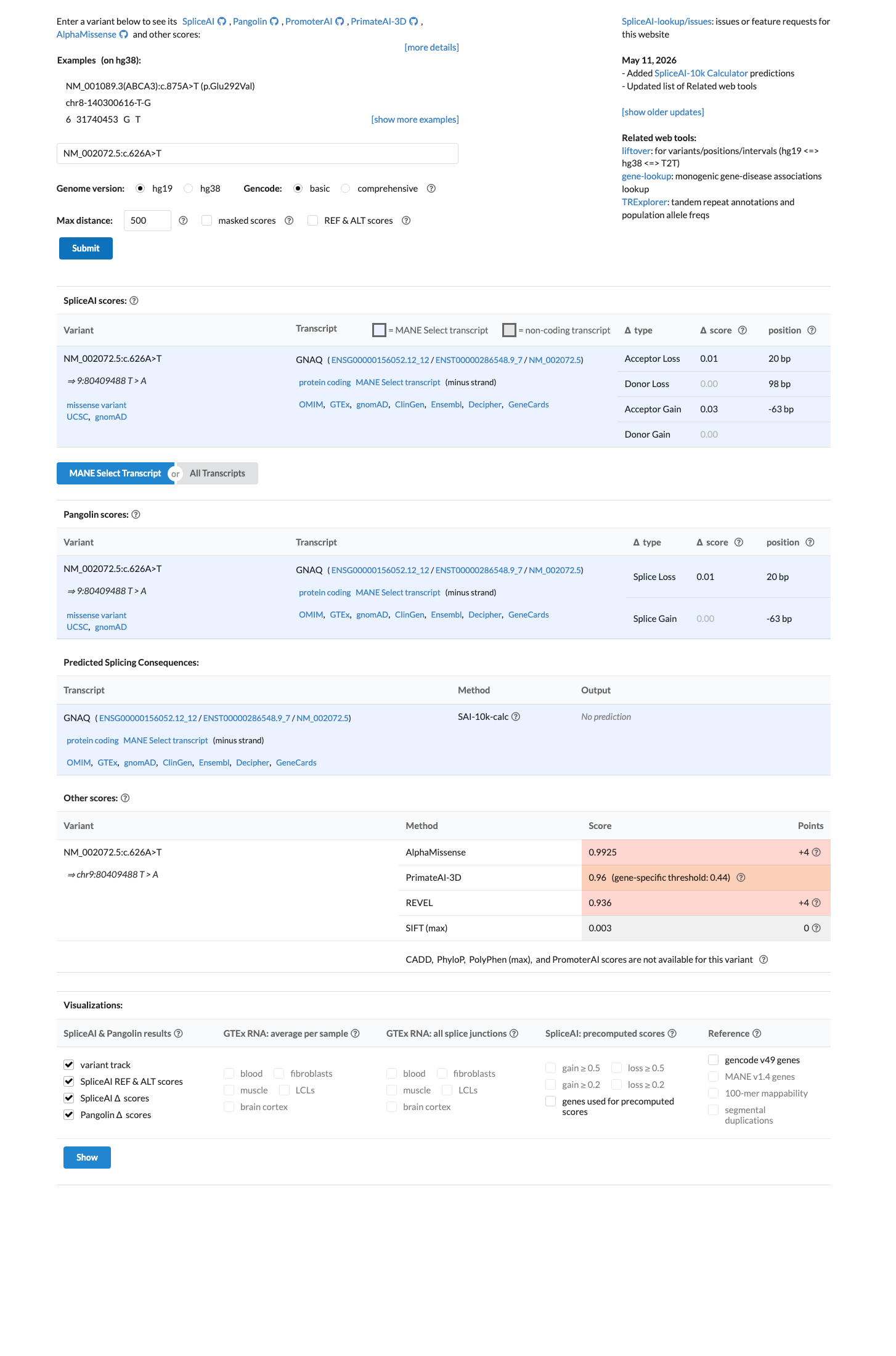
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.936. BayesDel score = 0.363052.
Functional
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Gain-of-function; curated oncogenicity label: Oncogenic.
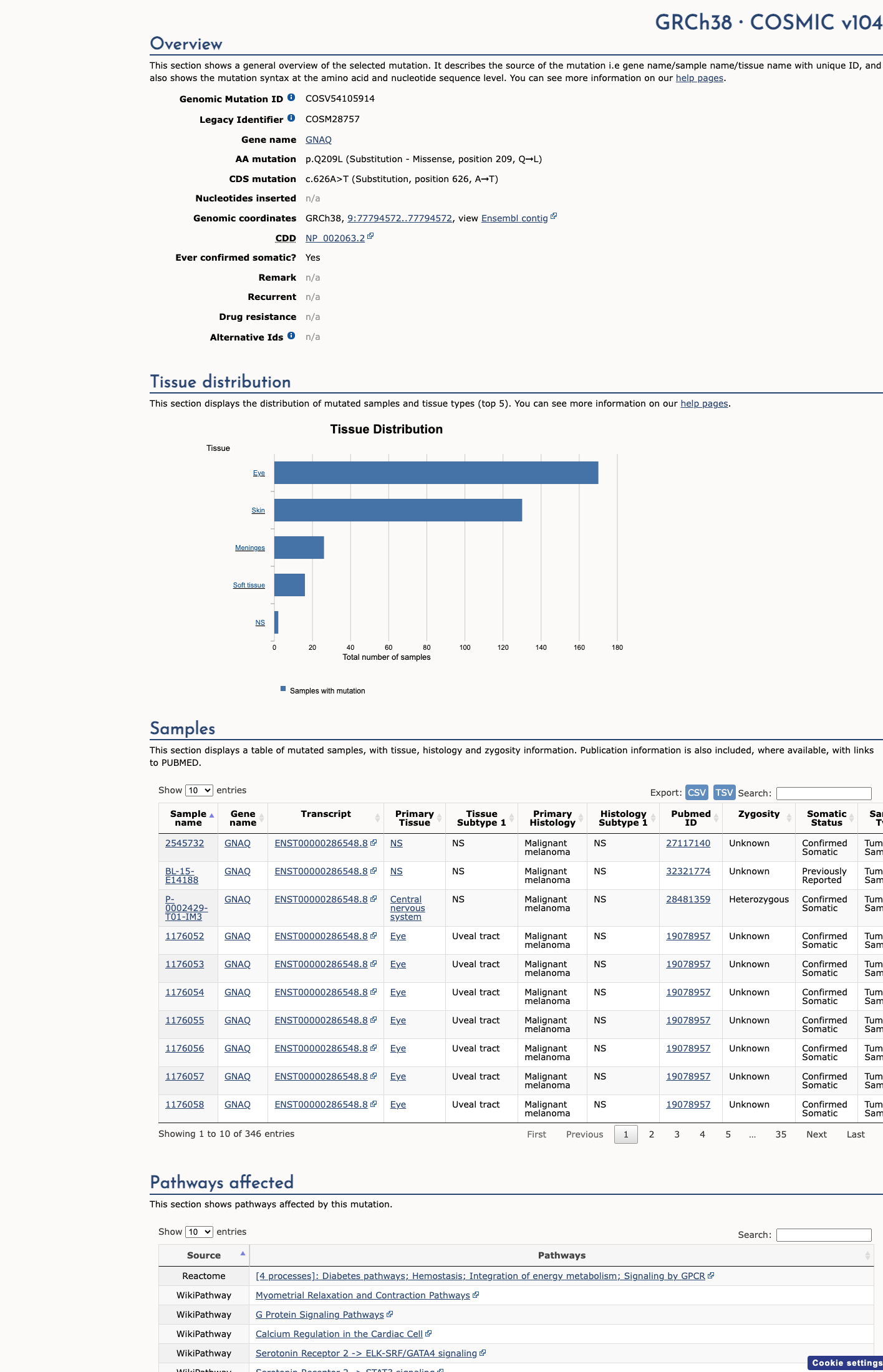
COSMIC
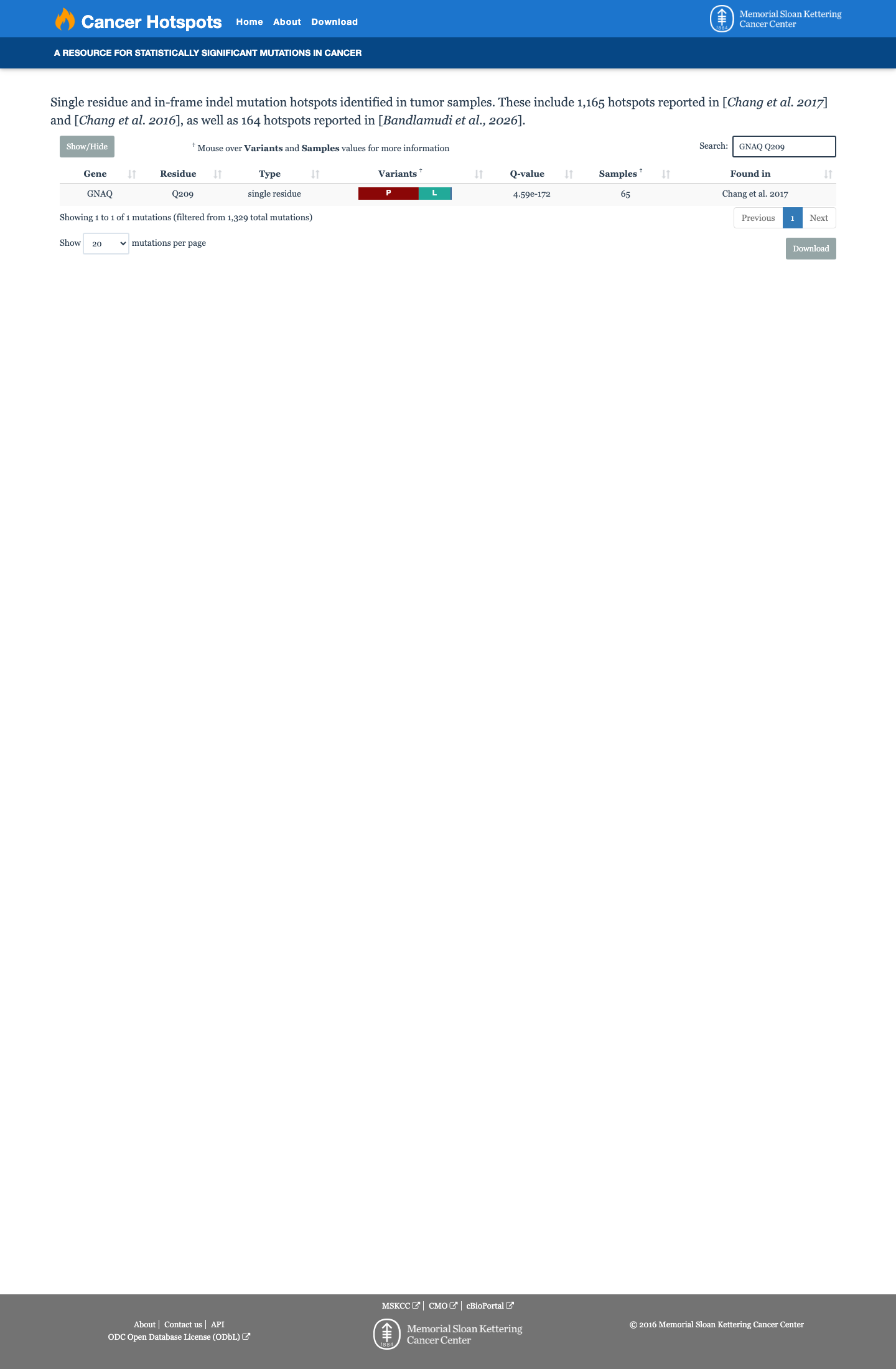
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV54105914, n = 346 times).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 2 further PMIDs triaged but not cited — see Sources & References.
Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells.
Found
Demonstrated that Q209L Gαq is GTPase-deficient and induces malignant transformation in NIH 3T3 cells.
Applied to
→PS3 supports · met
Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi.
Found
and PMID:19718445 confirm codon 209 as a recurrently mutated residue in human neoplasia.
Applied to
→PM1 supports · met
→PS3 supports · met
Mutational profile of GNAQQ209 in human tumors.
Found
and PMID:19718445 confirm codon 209 as a recurrently mutated residue in human neoplasia.
Applied to
→PM1 supports · met
→PM5 supports · met
→PS3 supports · met
GNAQ Q209R Mutations Are Highly Specific for Circumscribed Choroidal Hemangioma.
Found
Q209R identified as highly specific pathogenic variant in circumscribed choroidal hemangioma.
Applied to
→PM5 supports · met
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR