NM_007194.4:c.953G>A (p.Arg318His) is a missense variant in CHEK2 exon 9, located within the kinase domain. The variant has been observed at extremely low frequency in population databases (gnomAD v2.1: 13/282,864 alleles, AF=0.0046%; v4.1: 73/1,613,934 alleles, AF=0.0045%), consistent with PM2 at supporting level.1 Computational in silico tools uniformly predict a benign effect: REVEL score 0.061, BayesDel score -0.127872, and SpliceAI max delta 0.00, supporting BP4 at supporting level.2 Dong et al. (2003, PMID:12533788) identified this variant in 1 of 94 early-onset prostate cancer cases and 0 of 423 unaffected controls; it was not found in familial prostate cancer cases, and the single observation does not achieve statistical significance for PS4.3 ClinVar reports this variant as Uncertain Significance (15 clinical laboratories) and Likely Benign (1 laboratory). No expert panel has classified this variant.4 With PM2 (supporting pathogenic) and BP4 (supporting benign) applied, the evidence is balanced. The variant is classified as Variant of Uncertain Significance (VUS). Additional functional data (BS3/PS3) and case-control studies (PS4) are needed to resolve the classification.
CHEK2
Final classification
VUS
CHEK2 c.953G>A · p.Arg318His
CHEK2
NM_007194.4:c.953G>A (p.Arg318His) is a missense variant in CHEK2 exon 9, located within the kinase domain. The variant has been observed at extremely low frequency in population databases (gnomAD v2.1: 13/282,864 alleles, AF=0.0046%; v4.1: 73/1,613,934 alleles, AF=0.0045%), consistent with PM2 at supporting level.
Hereditary Breast, Ovarian and Pancreatic Cancer Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
CHEK2 c.953G>A
PM2 + BP4
→
VUS
Gene diagram
· NM_007194.4 · variants mapped to exon structure
CHEK2
NM_007194.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_007194.4:c.953G>A is present at extremely low frequency in population databases: gnomAD v2.1 AF=0.0046% (13/282,864 alleles, 0 homozygotes) and gnomAD v4.1 AF=0.0045% (73/1,613,934 alleles, 0 homozygotes). Both are below the 0.1% threshold for PM2.
gnomAD v2.1: AF=4.6e-05 (13/282864)v4.1: AF=4.5e-05 (73/1
✓
BP4
supporting
Benign
Multiple lines of computational evidence support a benign effect. REVEL score is 0.061 (below pathogenicity threshold), BayesDel score is -0.127872 (negative = benign), and SpliceAI predicts no splice impact (max delta = 0.00). All available in silico tools are concordant in predicting a neutral effect.
REVEL: 0.061BayesDel: -0.127872SpliceAI max delta: 0.00.
Assessed · not applied
Pathogenic
PS2
No de novo occurrence data (with confirmed paternity and maternity) was identified in the reviewed evidence for NM_007194.4:c.953G>A.
PS3
Well-established in vitro functional studies demonstrating a damaging effect on CHEK2 protein function were not confirmed in full-text review.
PS4
No statistically significant enrichment of NM_007194.4:c.953G>A was observed in affected individuals versus controls.
PM1
Although p.Arg318His resides within the CHEK2 kinase domain (a well-established functional domain spanning residues ~220-486), the specific residue is not a recognized mutational hotspot, and the variant has been observed in population databases at low frequency.
PM5
No pathogenic missense variant at the same amino acid residue (Arg318) with a different amino acid substitution was identified.
PM6
No de novo occurrence data (assumed or confirmed) was identified for NM_007194.4:c.953G>A.
PP1
No cosegregation data is available for this variant.
PP2
PP2 requires a gene with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP3
Multiple lines of in silico evidence predict a benign effect: REVEL score 0.061 (below the 0.5 pathogenicity threshold), BayesDel score -0.127872 (negative score indicates benign), and SpliceAI max delta 0.00 (no predicted splice impact).
PP4
No patient phenotype or family history data specific to the index case carrying NM_007194.4:c.953G>A was provided for review.
PP5
No reputable source has reported NM_007194.4:c.953G>A as pathogenic.
Benign
BA1
The maximum allele frequency in gnomAD v2.1 is 0.0046% (grpmax FAF 3.89e-05), far below the BA1 threshold of 1%.
BS1
The allele frequency in gnomAD is 0.0046% (v2.1) and 0.0045% (v4.1), far below the BS1 threshold of 0.3%.
BS2
While NM_007194.4:c.953G>A is observed in gnomAD (a general population cohort that includes healthy individuals), the frequency is extremely low (AF ~0.0046%) and does not constitute strong evidence for BS2, which requires observation in healthy adults at a frequency inconsistent with full penetrance for a dominant disorder.
BS3
Well-established functional studies showing no damaging effect on CHEK2 protein function were not confirmed in full-text review.
BS4
No cosegregation or nonsegregation data are available for NM_007194.4:c.953G>A.
BP1
BP1 applies to missense variants in genes where primarily truncating variants cause disease.
BP2
No phase data (in cis or in trans with a known pathogenic variant) is available for NM_007194.4:c.953G>A.
BP5
No data was available regarding an alternative molecular basis for disease in a proband carrying NM_007194.4:c.953G>A.
BP6
One clinical laboratory (Myriad Genetics, SCV005896018) classified this variant as Likely Benign, but this constitutes a single submitter.
N/A · 3
PVS1 · PS1 · BP7
Research & evidence
Population frequency

gnomAD v4.1
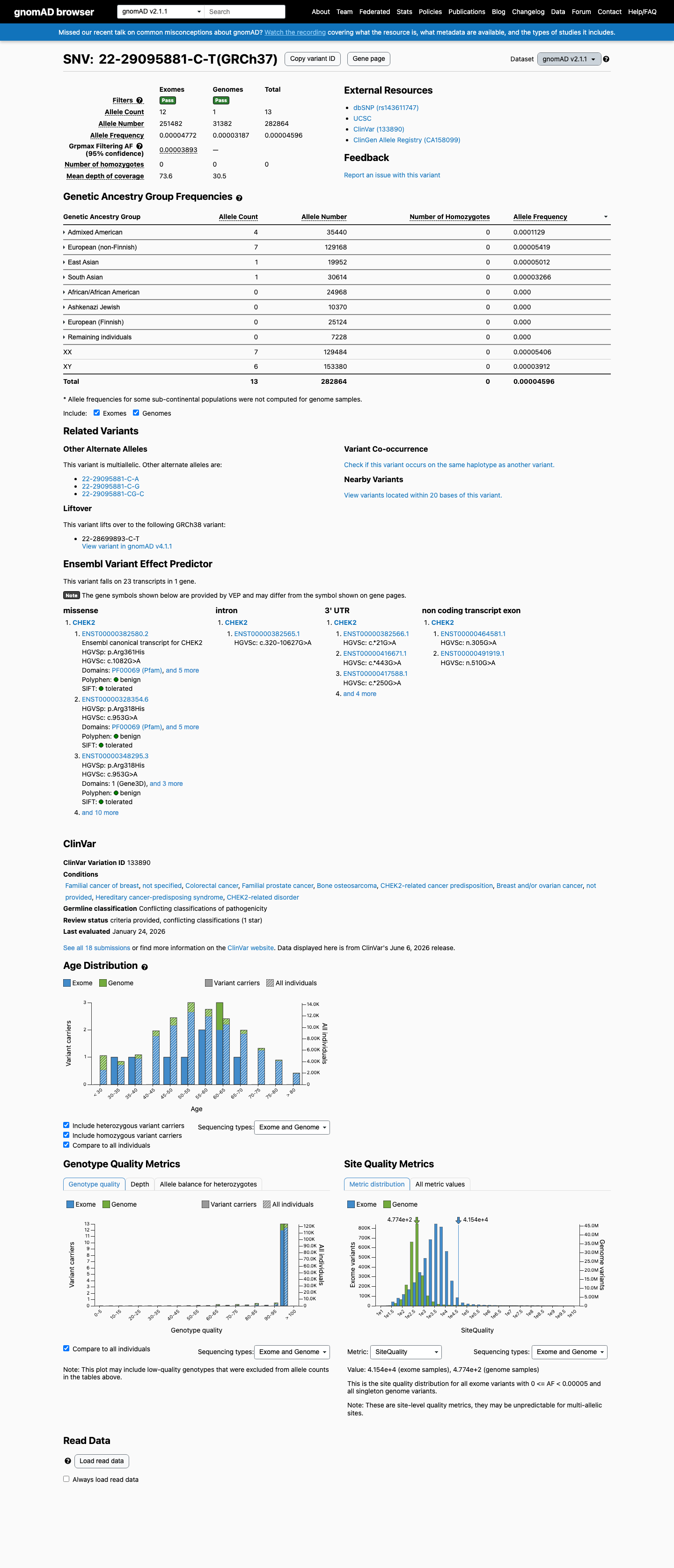
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 4.52311e-05; MAF= 0.00452%, 73/1613934 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000116733; MAF= 0.01167%, 7/59966 alleles, homozygotes = 0); grpmax FAF= 5.437e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 4.59585e-05; MAF= 0.00460%, 13/282864 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000112867; MAF= 0.01129%, 4/35440 alleles, homozygotes = 0); grpmax FAF= 3.893e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0045%
· 73 / 1,613,934
0 hom · FAF 0.0054%
0 hom · FAF 0.0054%
Admixed American 7 / 59,966 |
0.012% |
European (non-Finnish) 63 / 1,180,000 |
0.0053% |
East Asian 1 / 44,886 |
0.0022% |
African/African American 1 / 74,898 |
0.0013% |
South Asian 1 / 91,076 |
0.0011% |
+ 5 not observed (Remaining individuals, European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0046%
· 13 / 282,864
0 hom · FAF 0.0039%
0 hom · FAF 0.0039%
Admixed American 4 / 35,440 |
0.011% |
European (non-Finnish) 7 / 129,168 |
0.0054% |
East Asian 1 / 19,952 |
0.005% |
South Asian 1 / 30,614 |
0.0033% |
+ 4 not observed (African/African American, Ashkenazi Jewish, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
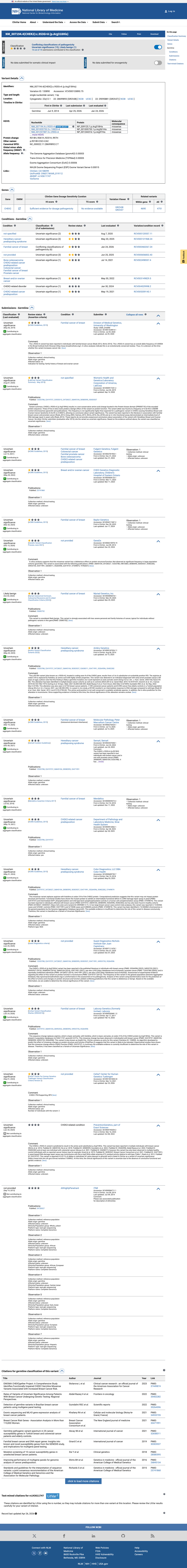
ClinVar
This variant has been reported in ClinVar as Uncertain significance (15 clinical laboratories) and as Likely benign (1 clinical laboratory). (ClinVarID = 133890)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.061. BayesDel score = -0.127872.
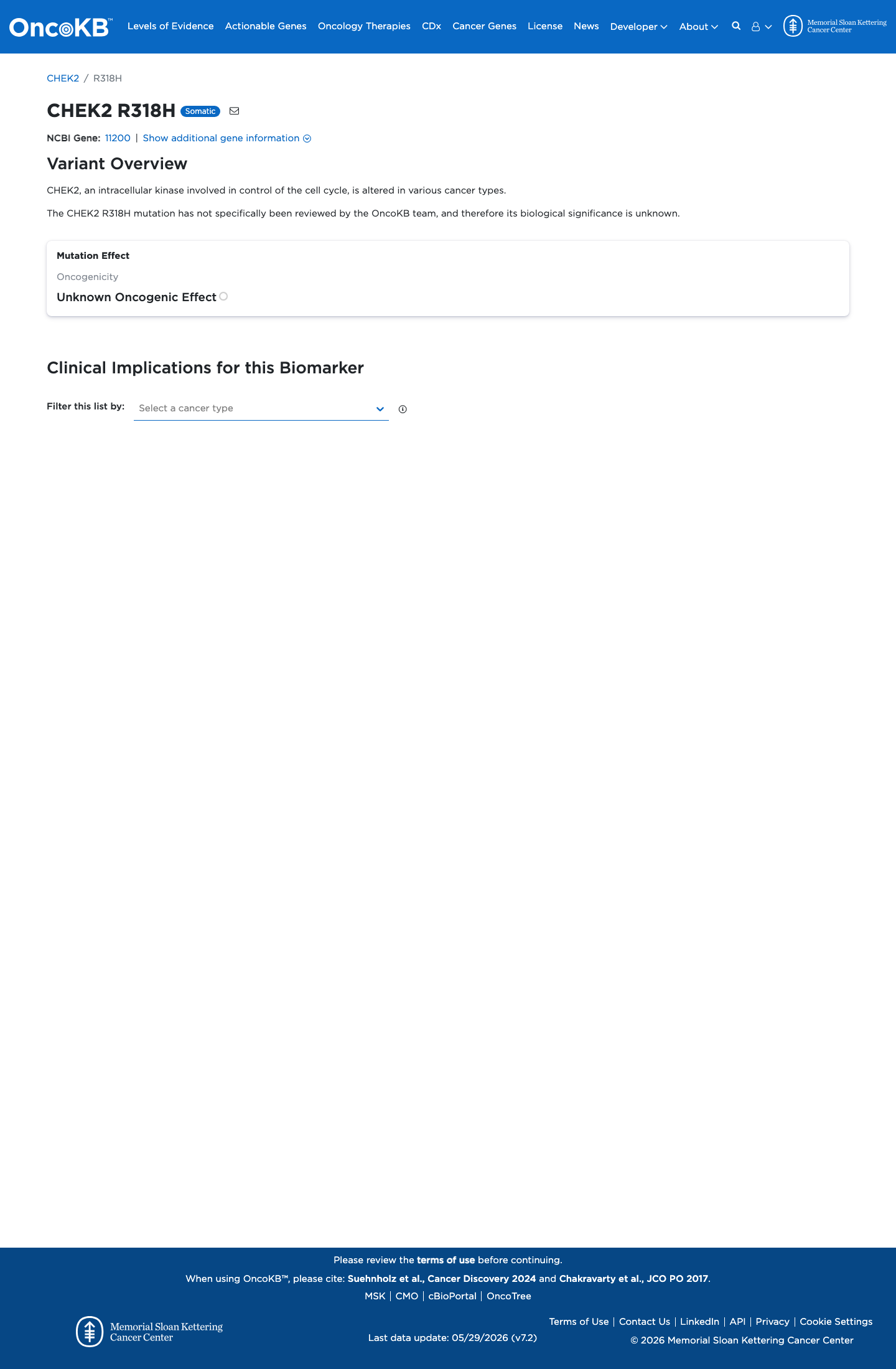
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. CHEK2, an intracellular kinase involved in control of the cell cycle, is altered in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
23555315 ↗
Genome-wide testing of putative functional exonic variants in relationship with breast and prostate cancer risk in a multiethnic population.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
26845104 ↗
Improving performance of multigene panels for genomic analysis of cancer predisposition.
CLINVAR
30303537 ↗
Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR