BA1 is met at stand-alone strength: gnomAD v4.1 grpmax filtering allele frequency is 0.027 (2.70%), far exceeding the VCEP threshold of ≥0.0022 (0.22%). The variant is observed in 6 homozygotes in v4.1 and 2 homozygotes in v2.1, and is not a known founder pathogenic variant.1 BP7 is met at supporting benign strength: the variant is an intronic duplication at positions -26 to -28 relative to exon 10, which is beyond the VCEP BP7 threshold of -21.2 BP4 is met at supporting benign strength: SpliceAI predicts no splicing impact (max delta score = 0.00), meeting the VCEP BP4_Supporting threshold of ≤0.1 for intronic variants.3 Per the InSiGHT MSH6 VCEP v2.0.0 combination rules, BA1 stand-alone alone is sufficient for a Benign classification (Rule 17).4
MSH6
Final classification
Benign
MSH6 c.4002-28_4002-26dup · p.?
MSH6
BA1 is met at stand-alone strength: gnomAD v4.1 grpmax filtering allele frequency is 0.027 (2.70%), far exceeding the VCEP threshold of ≥0.0022 (0.22%). The variant is observed in 6 homozygotes in v4.1 and 2 homozygotes in v2.1, and is not a known founder pathogenic variant.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MSH6 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule17 (1 Benign.Stand Alone) with applied criteria: BA1 stand-alone benign, BP4 supporting benign, BP7 supporting benign; maps to Benign.
Classification rationale
BA1BP4BP7
Benign
MSH6 c.4002-28_4002-26dup
BA1 + BP4 + BP7
→
Benign
Gene diagram
· NM_000179.3 · variants mapped to exon structure
MSH6
NM_000179.3
Fetching transcript structure from UCSC…
Applied criteria · 3 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
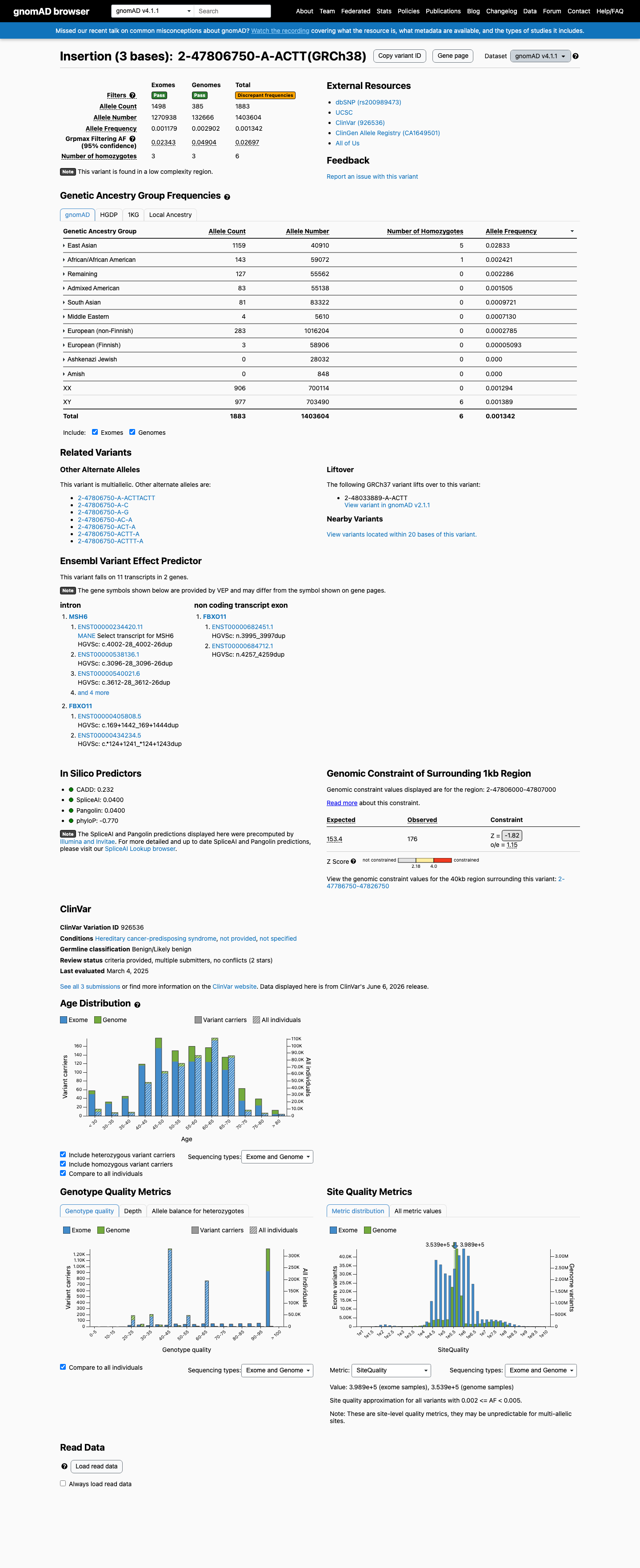
gnomAD v4.1
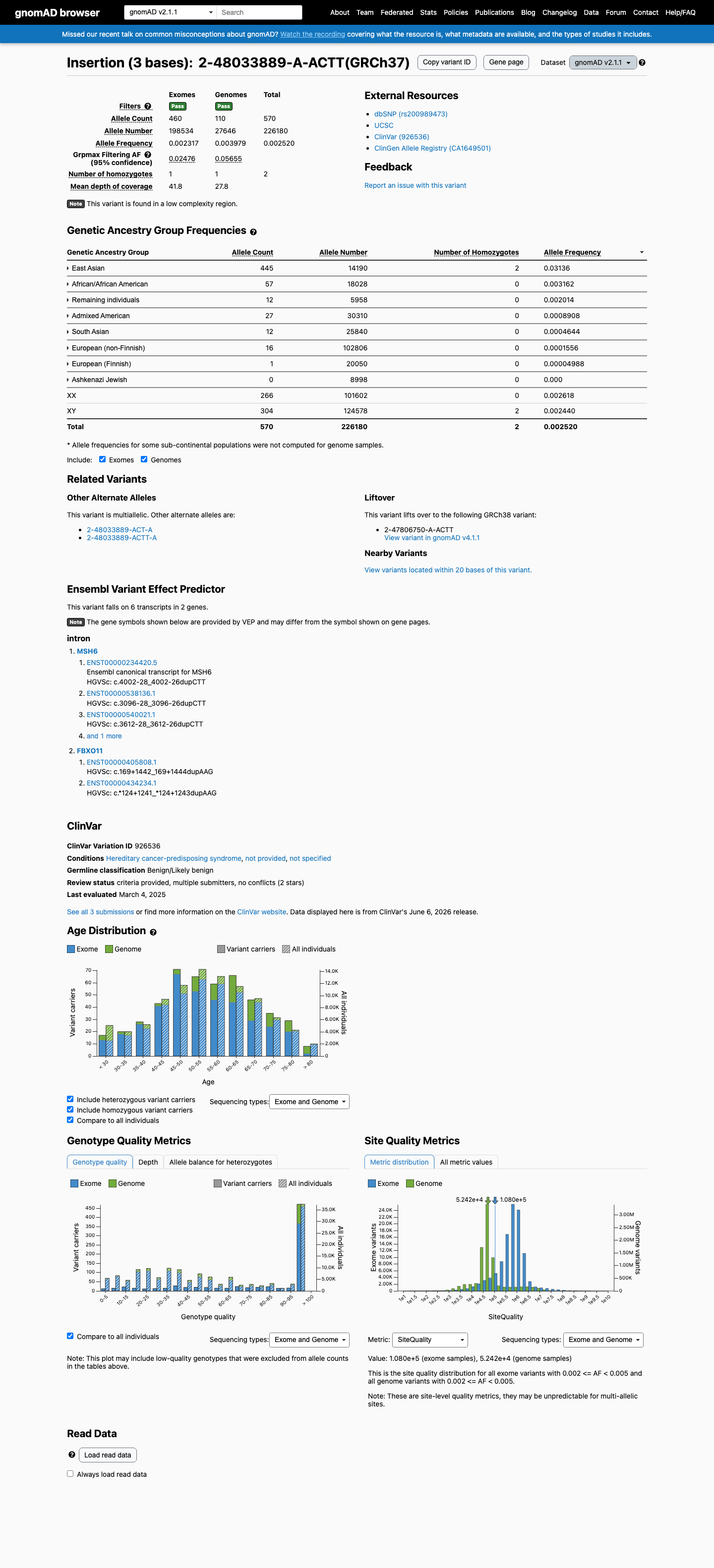
gnomAD v2.1
Population frequency
6
homozygotes observed in gnomAD v4.1. Healthy biallelic carriers are difficult to reconcile with a fully penetrant loss-of-function disease allele.
Overall AF
1883 / 1,403,604
0.13%
Highest · East Asian
2.8%
Homozygotes
6
grpmax FAF
2.7%
Allele frequency by ancestry — gnomAD v4.1
observed in 7 of 9 groups
| Ancestry | Allele count | Frequency | Homozygotes |
|---|---|---|---|
| East Asian | 1159 / 40,910 | 2.8% | 5 |
| African/African American | 143 / 59,072 | 0.24% | 1 |
| Admixed American | 83 / 55,138 | 0.15% | 0 |
| South Asian | 81 / 83,322 | 0.097% | 0 |
| Middle Eastern | 4 / 5,610 | 0.071% | 0 |
| European (non-Finnish) | 283 / 1,016,204 | 0.028% | 0 |
| European (Finnish) | 3 / 58,906 | 0.0051% | 0 |
| Amish | 0 / 848 | — | — |
| Ashkenazi Jewish | 0 / 28,032 | — | — |
“
This variant is present in gnomAD v4.1 (AF= 0.00134155; MAF= 0.13415%, 1883/1403604 alleles, homozygotes = 6) and has highest observed frequency in the East Asian population (AF= 0.0283305; MAF= 2.83305%, 1159/40910 alleles, homozygotes = 5); grpmax FAF= 0.0269749.
2
homozygotes observed in gnomAD v2.1. Healthy biallelic carriers are difficult to reconcile with a fully penetrant loss-of-function disease allele.
Overall AF
570 / 226,180
0.25%
Highest · East Asian
3.1%
Homozygotes
2
grpmax FAF
5.7%
Allele frequency by ancestry — gnomAD v2.1
observed in 7 of 8 groups
| Ancestry | Allele count | Frequency | Homozygotes |
|---|---|---|---|
| East Asian | 445 / 14,190 | 3.1% | 2 |
| African/African American | 57 / 18,028 | 0.32% | 0 |
| Remaining individuals | 12 / 5,958 | 0.2% | 0 |
| Admixed American | 27 / 30,310 | 0.089% | 0 |
| South Asian | 12 / 25,840 | 0.046% | 0 |
| European (non-Finnish) | 16 / 102,806 | 0.016% | 0 |
| European (Finnish) | 1 / 20,050 | 0.005% | 0 |
| Ashkenazi Jewish | 0 / 8,998 | — | — |
“
This variant is present in gnomAD v2.1 (AF= 0.00252012; MAF= 0.25201%, 570/226180 alleles, homozygotes = 2) and has highest observed frequency in the East Asian population (AF= 0.0313601; MAF= 3.13601%, 445/14190 alleles, homozygotes = 2); grpmax FAF= 0.0565538.
“
This variant is absent from gnomAD-Canada.
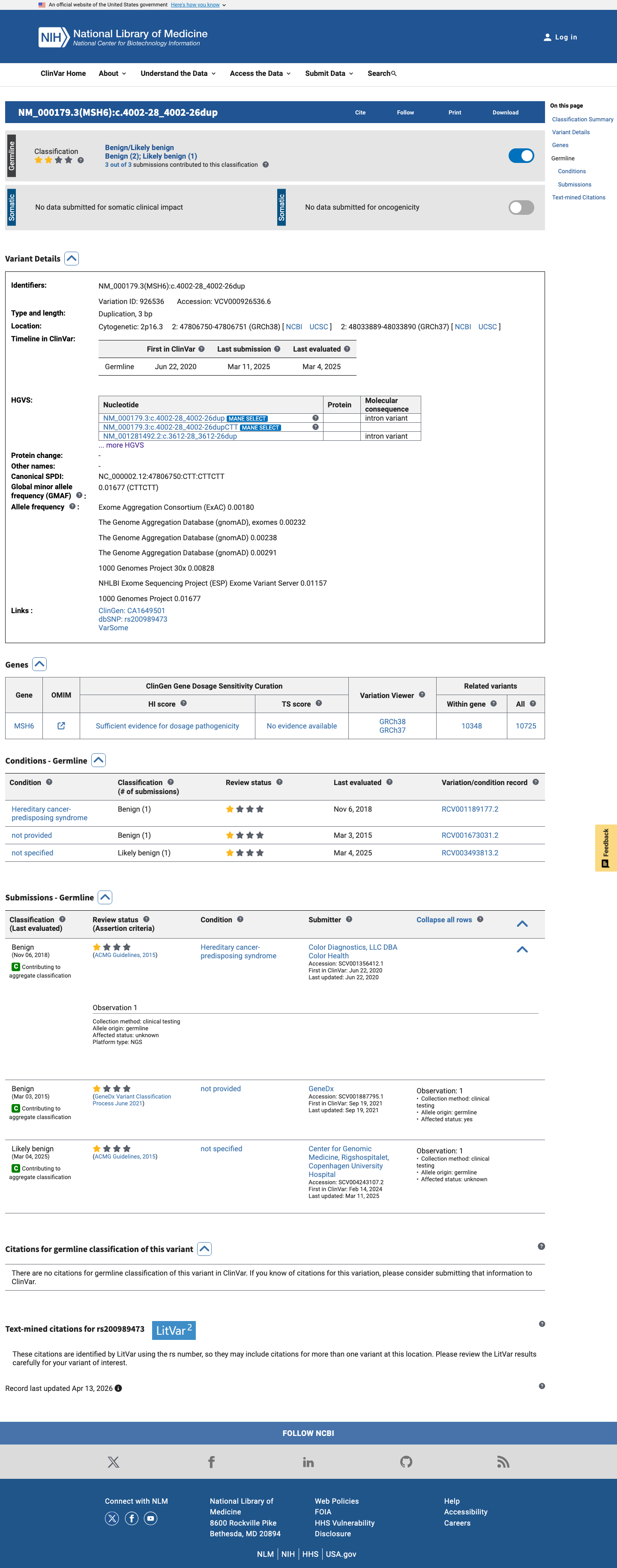
ClinVar
This variant has been reported in ClinVar as Benign (2 clinical laboratories) and as Likely benign (1 clinical laboratory). (ClinVarID = 926536)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · 2 PMIDs triaged · 2 high-priority
2papers screened
Papers triaged by theme: functional/splicing/segregation/case_observation. high_priority_papers include abstract snippets. Use these to support PS3/BS3/PS4/PP1/PP3/PP5.
25741868 ↗
functional
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
The American College of Medical Genetics and Genomics (ACMG) previously developed guidance for the interpretation of sequence variants.(1) In the past decade, sequencing technology has evolved rapidly with the advent of high-throughput next-generation sequencing. By adopting and leveraging next-generation sequencing, clinical laboratories are now performing an ever-increasing catalogue of genetic
BS3PP5PS3PS4
25394175 ↗
case observation
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
The practice guidelines of the American College of Medical Genetics and Genomics (ACMG) and the National Society of Genetic Counselors (NSGC) are developed by members of the ACMG and NSGC to assist medical geneticists, genetic counselors, and other health-care providers in making decisions about appropriate management of genetic concerns, including access to and/or delivery of services. Each pract
PP5PS4
Sources & reference links