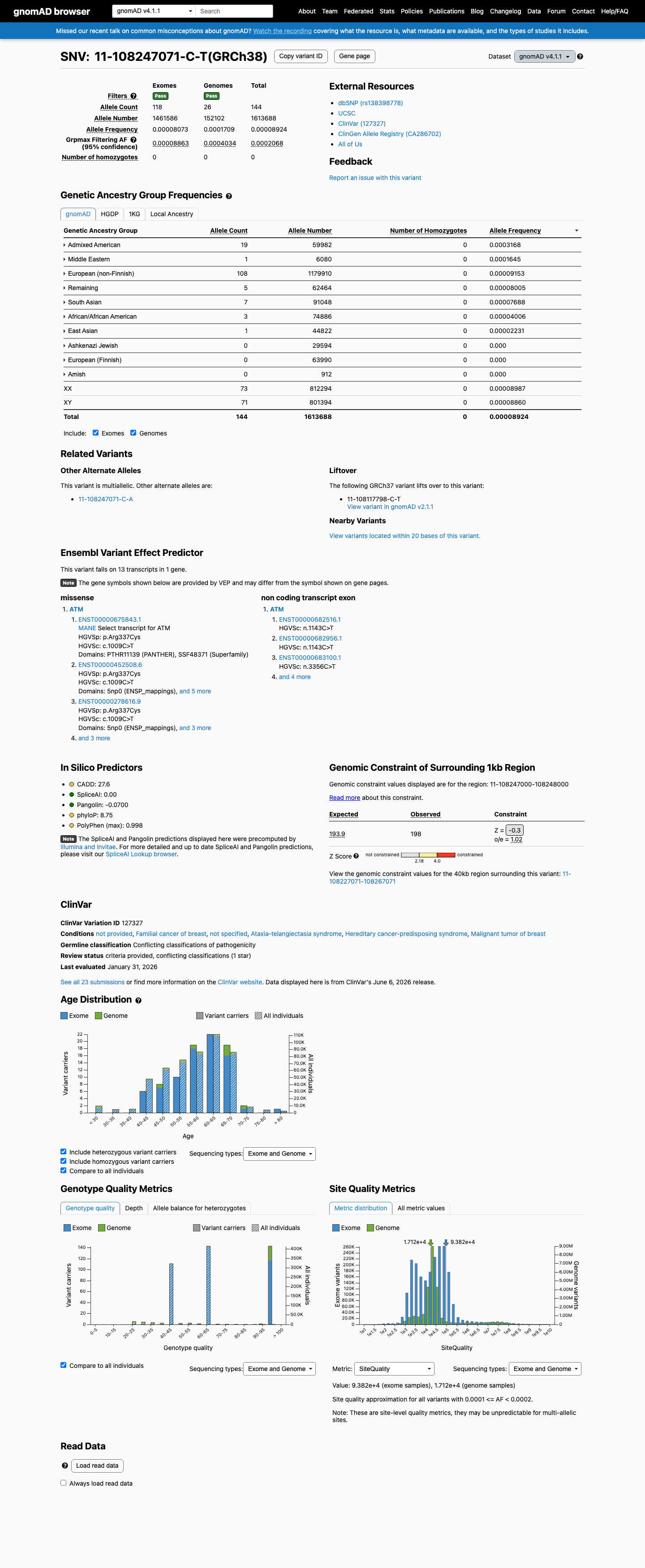
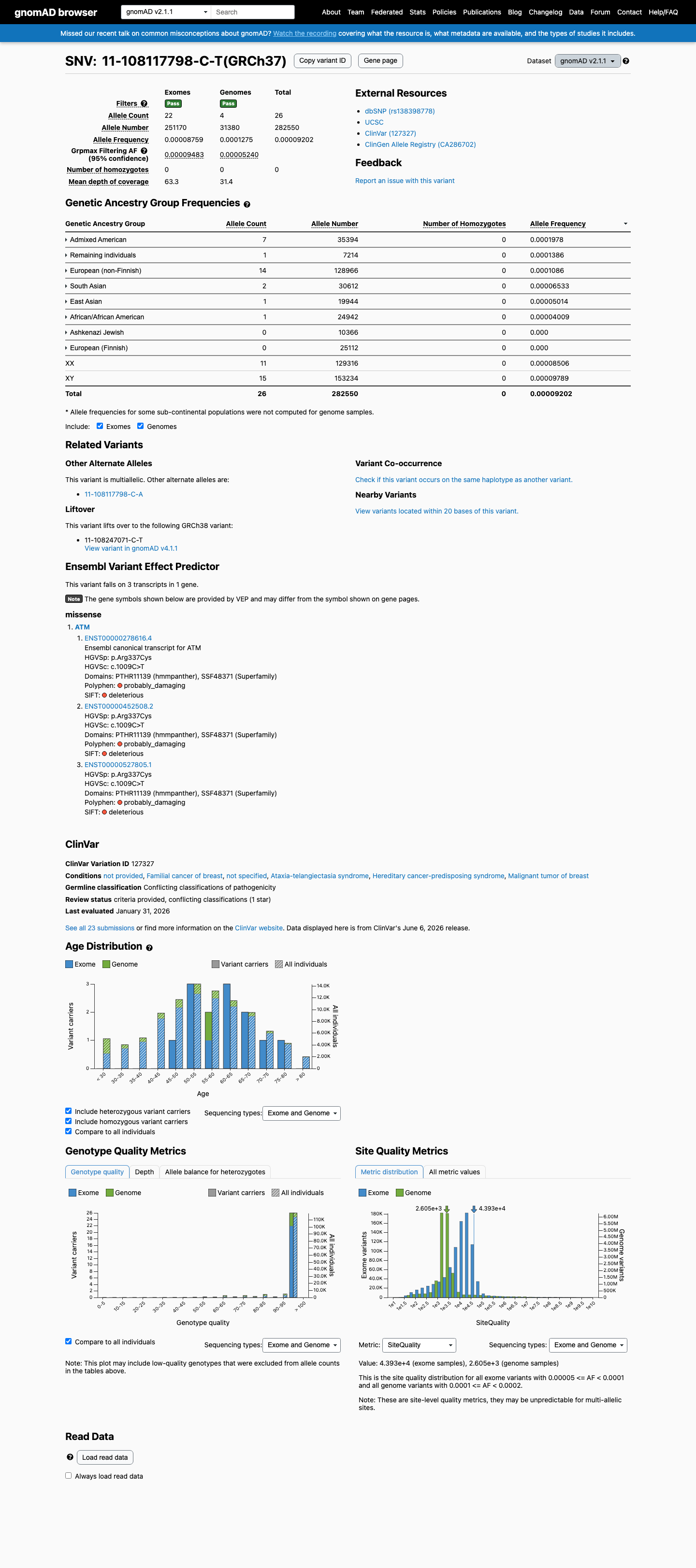
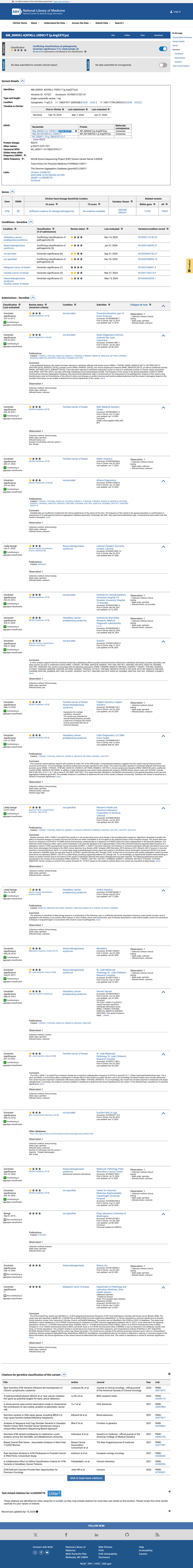
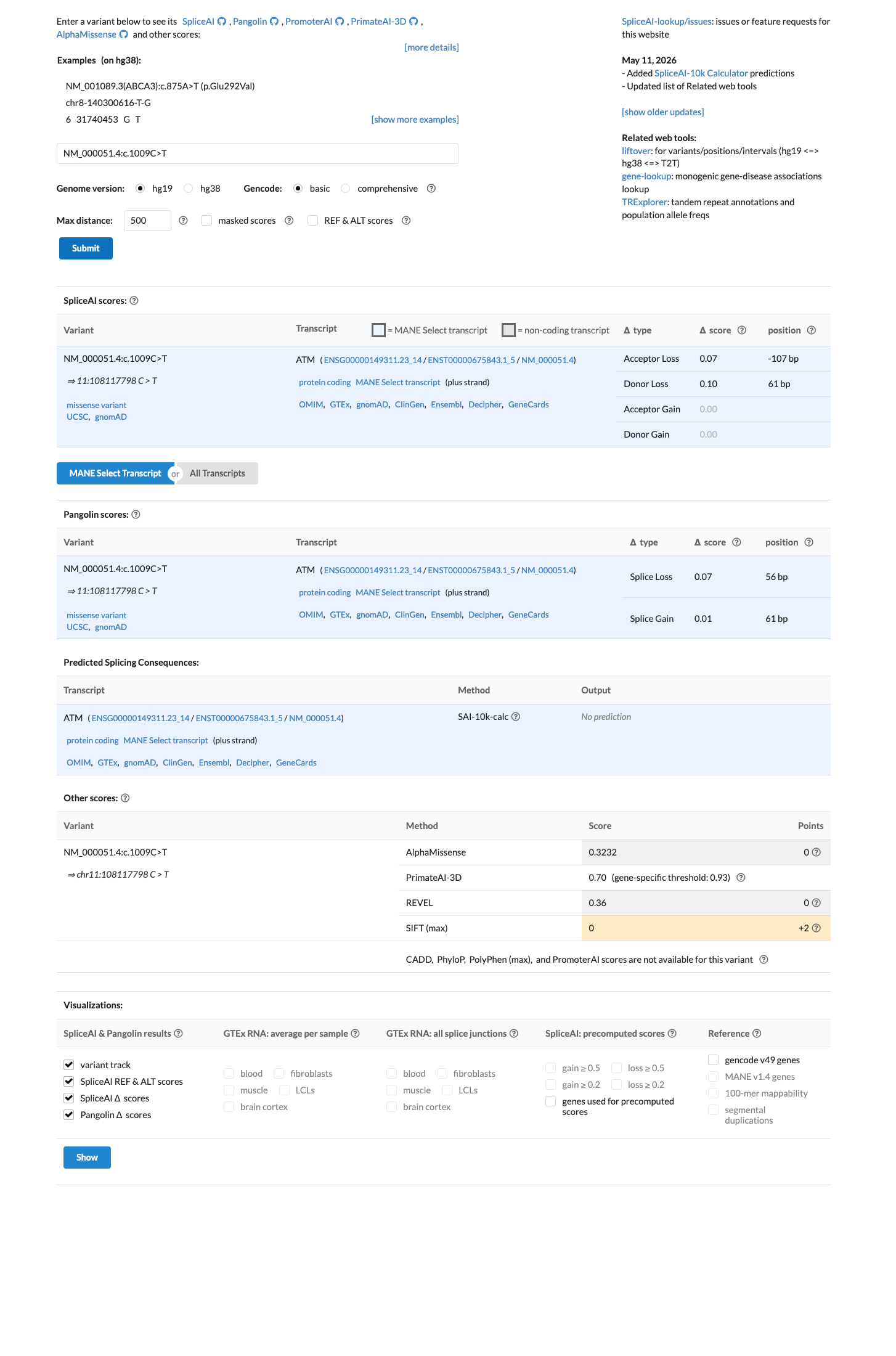
NM_000051.4:c.1009C>T (p.Arg337Cys) is a missense variant in ATM exon 8. This variant is present in gnomAD v4.1 at an overall allele frequency of 0.00892% (144/1,613,688 alleles, 0 homozygotes) with a grpmax filtering allele frequency of 0.021% in the Admixed American population.1 This variant does not meet PM2_Supporting (frequency ≤0.001% required), BS1 (>0.05% required), or BA1 (>0.5% required) under ATM VCEP population thresholds.2 This variant has been reported in ClinVar (Variation ID 127327) as Uncertain Significance by 18 clinical laboratories and as Likely Benign by 3 clinical laboratories; no submitter classifies it as pathogenic.3 The variant has been reported 80 times in somatic cancers (COSMIC COSV53723836) and OncoKB classifies it as Likely Oncogenic with a loss-of-function effect in the somatic context.4 Computational predictors do not support a deleterious effect: REVEL score is 0.36 (VCEP PP3 threshold >0.7333, BP4 threshold ≤0.249), SpliceAI max delta is 0.10, and BayesDel is 0.067.5 The ATM VCEP functional dataset (Suppl_TableS1, PMID 40580951) classifies this variant as 'Functional' (combined score -0.65, high confidence) based on integrated functional and evolutionary scores, meeting BS3_Supporting under VCEP rules.6 PVS1, PS1, PS2, PM1, PM5, PM6, PP2, PP4, PP5, BS2, BS4, BP1, BP5, BP6, and BP7 are not applicable under ATM VCEP v1.5.0 for this variant type.7 No case-control study demonstrates enrichment of this variant in affected individuals (PS4 not met), and no segregation data are available (PP1 not assessed).8 With only BS3_Supporting met and no pathogenic criteria fulfilled, the evidence is insufficient to classify this variant as either likely benign or likely pathogenic; the variant remains a Variant of Uncertain Significance under the ATM VCEP framework.9
ATM
Final classification
VUS
ATM c.1009C>T · p.Arg337Cys
ATM
NM_000051.4:c.1009C>T (p.Arg337Cys) is a missense variant in ATM exon 8.
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework was evaluated deterministically with applied criteria: BS3 supporting benign; no rule matched the adjudicated criteria.
Classification rationale
BS3
VUS
ATM c.1009C>T
BS3
→
VUS
4
oncokb ↗
5
revelspliceai ↗bayesdel
6
vcep_suppl_tables1_pmid_40580951vcep_clingen_hbop_atm_supplementary_tables_1_and_2_v1
7
cspec ↗
9
cspec ↗
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 10 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BS3
supporting
Benign
The ATM VCEP functional dataset (Suppl_TableS1, PMID 40580951) classifies c.1009C>T (p.Arg337Cys) as 'Functional' (combined score -0.65, function score 31, high confidence), indicating retention of ATM activity. Under VCEP BS3 rules, a variant that rescues ATM-specific functional features (e.g., kinase activity) meets BS3_Supporting. Functional studies in the Barone et al. 2009 and Mitui et al. 2009 datasets demonstrate retained ATM kinase activity for this variant.
VCEP functional dataset classifies variant as 'Functional' with high confidenceATM kinase activity assay references (Barone 2009Mitui 2009) included in VCEP supplementary tables.
Assessed · not applied
Pathogenic
PS1
No missense variant at codon 337 (Arg337) has been classified as pathogenic or likely pathogenic by VCEP criteria.
PS3
The ATM VCEP functional dataset (Suppl_TableS1, PMID 40580951) classifies c.1009C>T (p.Arg337Cys) as 'Functional' with a combined score of -0.65, indicating retention of ATM activity.
PS4
No case-control study demonstrates statistically significant enrichment of this variant in affected individuals.
PM2
gnomAD v4.1 allele frequency is 0.00892% (144/1,613,688 alleles), exceeding the ATM VCEP PM2_Supporting threshold of ≤0.001%.
PP1
No published cosegregation data are available for this variant in families with ATM-related conditions.
PP3
REVEL score 0.36 does not meet the ATM VCEP PP3 threshold (>0.7333) for missense variants.
Benign
BA1
gnomAD v4.1 grpmax filtering allele frequency is 0.021% (2.07×10⁻⁴), well below the ATM VCEP BA1 threshold of >0.5%.
BS1
gnomAD v4.1 grpmax filtering allele frequency is 0.021% (2.07×10⁻⁴), below the ATM VCEP BS1 threshold of >0.05%.
BP2
No confirmed observation of this variant in trans with a pathogenic or likely pathogenic ATM variant in an unaffected individual aged ≥18 years has been verified.
BP4
REVEL score 0.36 exceeds the ATM VCEP BP4 threshold (≤0.249) for missense variants.
N/A · 14
PVS1 · PS2 · PM1 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP5 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 8.92366e-05; MAF= 0.00892%, 144/1613688 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000316762; MAF= 0.03168%, 19/59982 alleles, homozygotes = 0); grpmax FAF= 0.00020684.
v2.1
This variant is present in gnomAD v2.1 (AF= 9.20191e-05; MAF= 0.00920%, 26/282550 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000197774; MAF= 0.01978%, 7/35394 alleles, homozygotes = 0); grpmax FAF= 9.483e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0089%
· 144 / 1,613,688
0 hom · FAF 0.021%
0 hom · FAF 0.021%
Admixed American 19 / 59,982 |
0.032% |
Middle Eastern 1 / 6,080 |
0.016% |
European (non-Finnish) 108 / 1,179,910 |
0.0092% |
Remaining individuals 5 / 62,464 |
0.008% |
South Asian 7 / 91,048 |
0.0077% |
African/African American 3 / 74,886 |
0.004% |
East Asian 1 / 44,822 |
0.0022% |
+ 3 not observed (European (Finnish), Amish, Ashkenazi Jewish)
gnomAD v2.1
0.0092%
· 26 / 282,550
0 hom · FAF 0.0095%
0 hom · FAF 0.0095%
Admixed American 7 / 35,394 |
0.02% |
Remaining individuals 1 / 7,214 |
0.014% |
European (non-Finnish) 14 / 128,966 |
0.011% |
South Asian 2 / 30,612 |
0.0065% |
East Asian 1 / 19,944 |
0.005% |
African/African American 1 / 24,942 |
0.004% |
+ 2 not observed (Ashkenazi Jewish, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (18 clinical laboratories) and as Likely benign (3 clinical laboratories). (ClinVarID = 127327)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.10). REVEL score = 0.36. BayesDel score = 0.0673325.
Functional
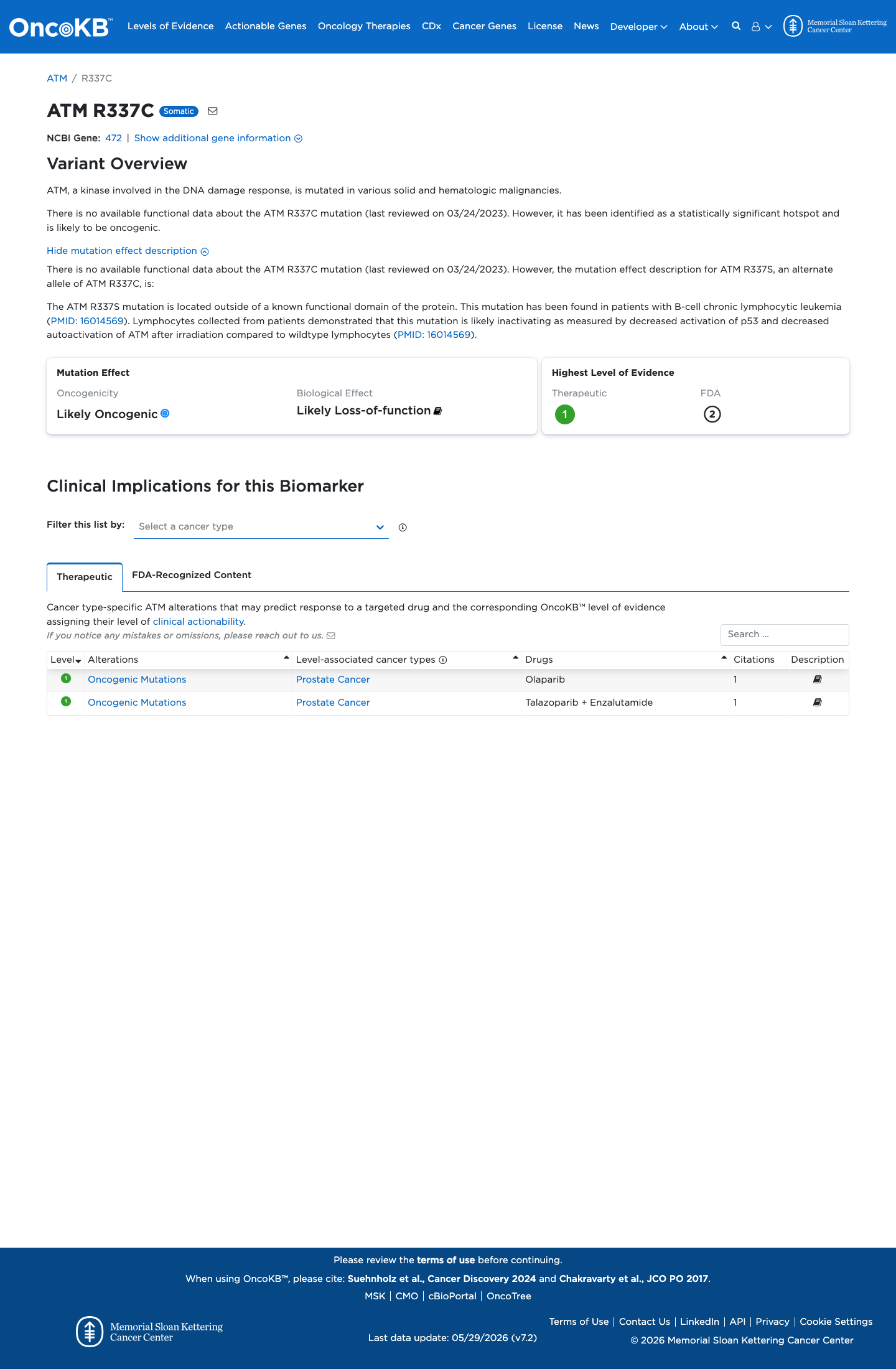
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
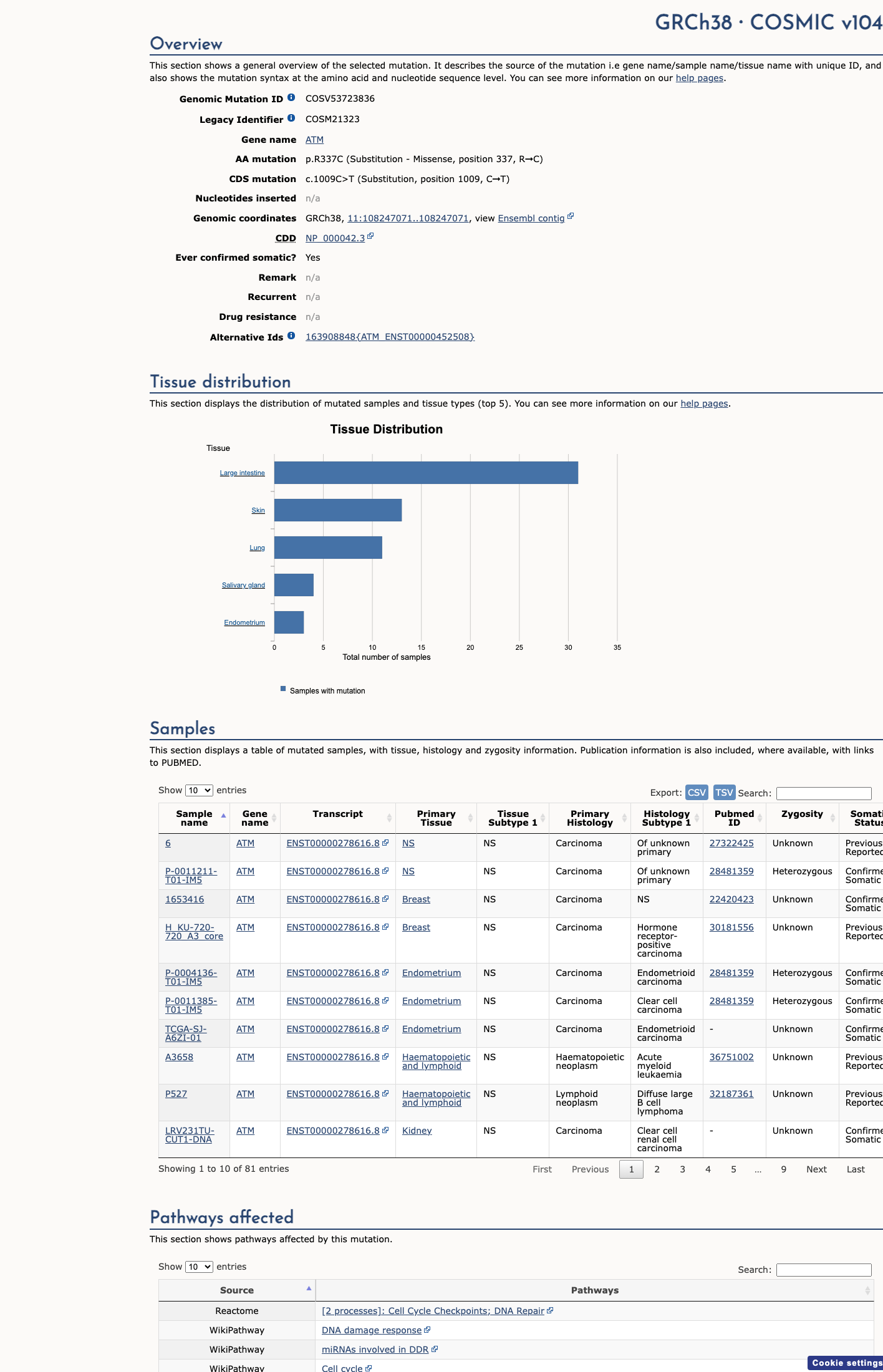
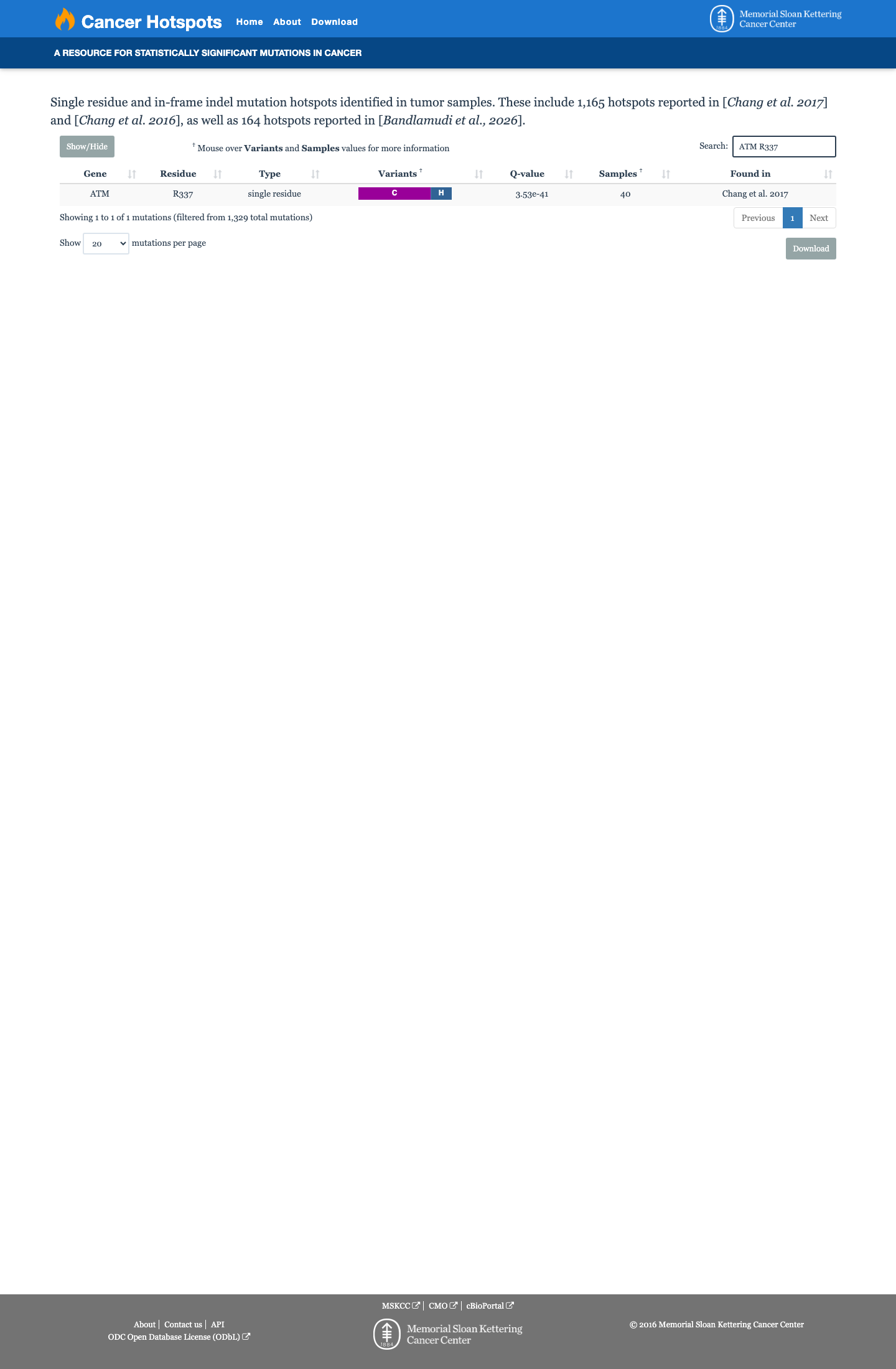
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53723836, n = 80 times).
Hotspots
This variant lies in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
16014569 ↗
Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL.
ONCOKB
19781682 ↗
Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer.
CLINVAR
20305132 ↗
Radiation exposure, the ATM Gene, and contralateral breast cancer in the women's environmental cancer and radiation epidemiology study.
CLINVAR
22529920 ↗
Computational refinement of functional single nucleotide polymorphisms associated with ATM gene.
CLINVAR
23555315 ↗
Genome-wide testing of putative functional exonic variants in relationship with breast and prostate cancer risk in a multiethnic population.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
27844328 ↗
Molecular Profiling of Thymoma and Thymic Carcinoma: Genetic Differences and Potential Novel Therapeutic Targets.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR