NM_002755.3:c.146G>A (p.Arg49His) in MAP2K1 is a missense variant located within the VCEP-designated critical functional domain spanning amino acids 43-61, satisfying PM1 at Moderate strength.1 A confirmed de novo occurrence of this variant was reported in a patient with cardio-facio-cutaneous syndrome, with both maternity and paternity confirmed, satisfying PS2 at Moderate strength (1 point under VCEP scoring). The REVEL in silico score is 0.707, meeting the VCEP PP3 threshold of ≥0.7 for missense variants at Supporting strength.2 This variant is present at extremely low frequency in gnomAD (AF ~0.0004%), far below both the BA1 (≥0.05%) and BS1 (≥0.025%) thresholds, and present in only 1 allele in v2.1 and 5 alleles in v4.1.3 No alternative pathogenic missense change at codon 49 was identified (PM5 not met), and the variant is not absent from gnomAD controls (PM2 not met per VCEP requirement for complete absence).4 PP2 (missense constraint z-score) remains unassessed pending gnomAD constraint data retrieval. If the MAP2K1 missense z-score exceeds 3.09, PP2_Supporting would also be met.5 ClinVar lists this variant as Uncertain significance (1 submitter, SCV003504233, Labcorp Genetics). No expert panel classification is available.6 Under the RASopathy VCEP v2.3.0 combination rules, the current met criteria (PM1_Moderate + PS2_Moderate + PP3_Supporting) do not satisfy any rule for Likely Pathogenic or Pathogenic classification. If PP2_Supporting is also met, Rule14 (2 Moderate + ≥2 Supporting) would be satisfied, yielding Likely Pathogenic.7
MAP2K1
Final classification
VUS
MAP2K1 c.146G>A · p.Arg49His
MAP2K1
NM_002755.3:c.146G>A (p.Arg49His) in MAP2K1 is a missense variant located within the VCEP-designated critical functional domain spanning amino acids 43-61, satisfying PM1 at Moderate strength.
ClinGen RASopathy Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MAP2K1 Version 2.3.0 v2.3.0 criteria-combination framework was evaluated deterministically with applied criteria: PS2 moderate, PM1 moderate, PP3 supporting; no rule matched the adjudicated criteria.
Classification rationale
PS2PM1PP3
VUS
MAP2K1 c.146G>A
PS2 + PM1 + PP3
→
VUS
Gene diagram
· NM_002755.3 · variants mapped to exon structure
MAP2K1
NM_002755.3
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 15 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PS2
moderate
review
Pathogenic
A confirmed de novo occurrence of NM_002755.3:c.146G>A (p.Arg49His) was reported in a patient with cardio-facio-cutaneous syndrome; both parents were tested and the variant was absent in both, satisfying 1 point under the RASopathy VCEP PS2 point-based scoring system (Moderate strength).
Exploratory evidence retrieval identified Rodriguez-Viciana et al. (2006DOI:10.1038/ng1746PMID:16439621) reporting a de novo germline c.146G>A mutation in a CFC patient with both maternity and paternity confirmed. This qualifies for 1 point = PS2_Moderate under VCEP rules.
✓
PM1
moderate
Pathogenic
The variant affects amino acid position 49 (p.Arg49His), which lies within the VCEP-designated critical functional domain spanning amino acids 43-61 of MAP2K1. This region is specified in the RASopathy VCEP supplementary table as a well-established functional domain eligible for PM1 at Moderate strength.
RASopathy VCEP v2.3.0 PM1 rule: 'Applicable only to critical and well-established functional domains available in the supplementary table (AA 43-61AA 124-134).' Residue 49 falls within AA 43-61.
✓
PP3
supporting
Pathogenic
The REVEL score for NM_002755.3:c.146G>A (p.Arg49His) is 0.707, which meets the RASopathy VCEP PP3 threshold of REVEL ≥ 0.7 for missense variants. SpliceAI predicts no splice impact (max delta = 0.00), so the in silico evidence derives entirely from the REVEL missense prediction.
REVEL = 0.707 (≥0.7 threshold met). SpliceAI max delta = 0.00 (no splice impact). BayesDel = 0.139 (not incorporated in VCEP PP3 rule).
Assessed · not applied
Pathogenic
PS1
No previously established pathogenic variant with the same amino acid change (p.Arg49His) from a different nucleotide change has been identified in ClinVar or the literature for MAP2K1 or the analogous MAP2K2 residue.
PS3
No evidence that p.Arg49His has been evaluated in VCEP-approved functional assays with a demonstrated damaging effect.
PS4
Insufficient case-control evidence to apply PS4 under the VCEP point-based system.
PM2
The RASopathy VCEP PM2 rule requires the variant to be absent from gnomAD controls.
PM5
No alternative pathogenic or likely pathogenic missense variant at codon 49 of MAP2K1 (or the analogous MAP2K2 residue) was identified.
PM6
The single de novo observation of c.146G>A (PMID:16439621) had both maternity and paternity confirmed, qualifying it as PS2 rather than PM6.
PP1
No evidence of co-segregation of c.146G>A in multiple affected family members was identified.
PP2
The RASopathy VCEP PP2 rule requires a missense z-score >3.09 in gnomAD for MAP2K1.
Benign
BA1
The RASopathy VCEP BA1 threshold is a gnomAD filtering allele frequency ≥ 0.05%.
BS1
The RASopathy VCEP BS1 threshold is a gnomAD filtering allele frequency ≥ 0.025%.
BS2
No evidence that NM_002755.3:c.146G>A has been observed in a healthy adult individual without a RASopathy phenotype.
BS4
No evidence of non-segregation of c.146G>A in affected family members has been reported.
BP2
No evidence that c.146G>A has been observed in trans with another pathogenic MAP2K1 variant or that an alternative molecular cause in MAP2K1 explains the phenotype in a patient carrying this variant.
BP4
The RASopathy VCEP BP4 threshold for missense variants is REVEL ≤ 0.3.
BP5
No evidence of an alternative molecular cause in a different gene that fully explains the RASopathy phenotype in a patient carrying c.146G>A.
N/A · 7
PVS1 · PP4 · PP5 · BS3 · BP1 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
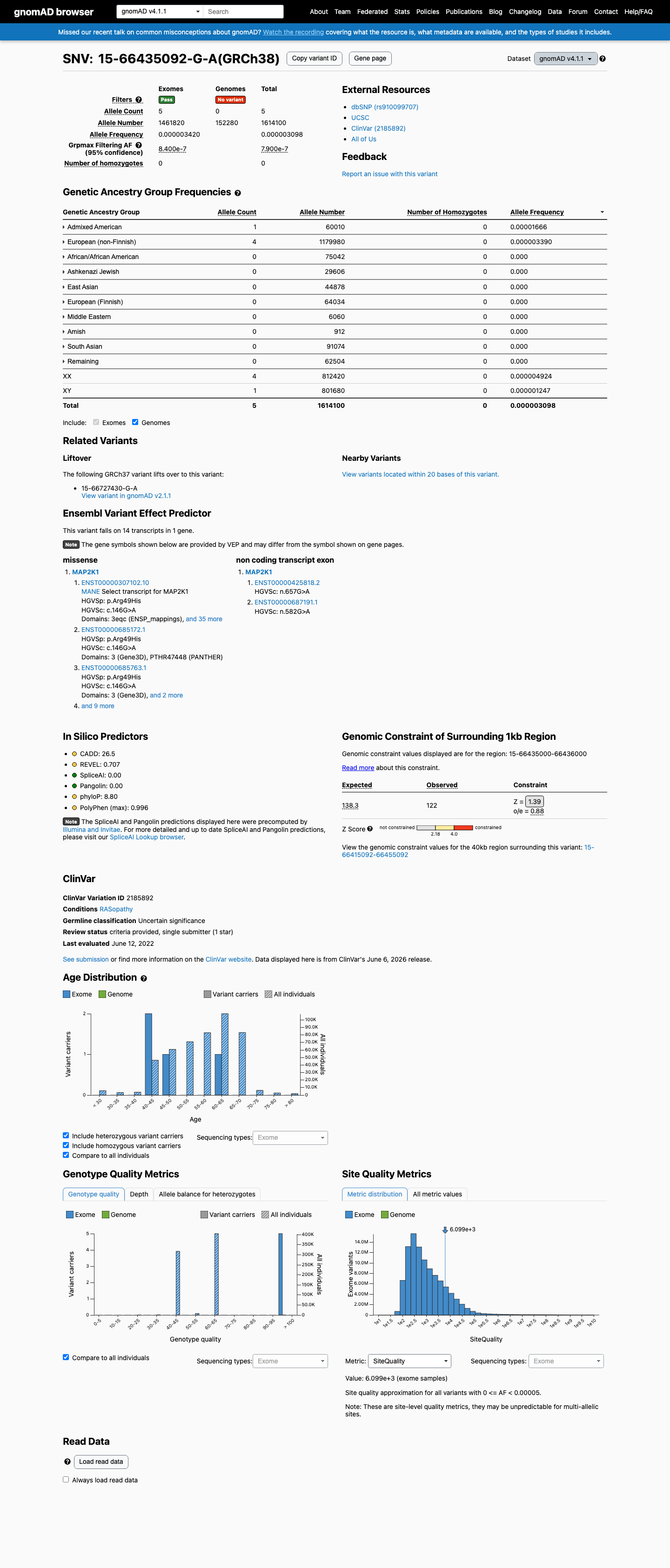
This variant is present in gnomAD v4.1 (AF= 3.0977e-06; MAF= 0.00031%, 5/1614100 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 1.66639e-05; MAF= 0.00167%, 1/60010 alleles, homozygotes = 0); grpmax FAF= 7.9e-07.
v2.1
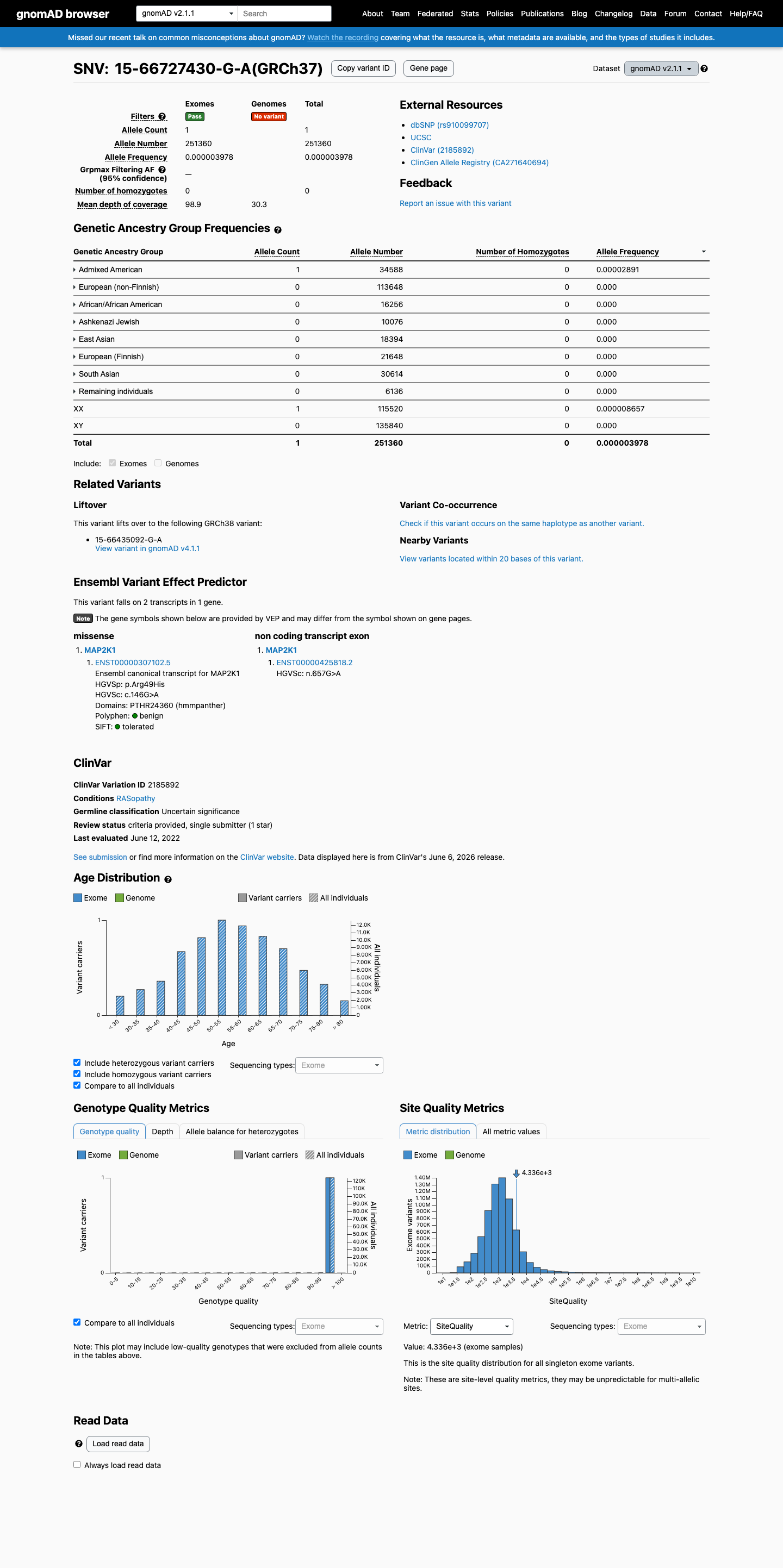
This variant is present in gnomAD v2.1 (AF= 3.97836e-06; MAF= 0.00040%, 1/251360 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 2.89118e-05; MAF= 0.00289%, 1/34588 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00031%
· 5 / 1,614,100
0 hom · FAF 7.9e-05%
0 hom · FAF 7.9e-05%
Admixed American 1 / 60,010 |
0.0017% |
European (non-Finnish) 4 / 1,179,980 |
0.00034% |
+ 8 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 251,360
0 hom
0 hom
Admixed American 1 / 34,588 |
0.0029% |
+ 7 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
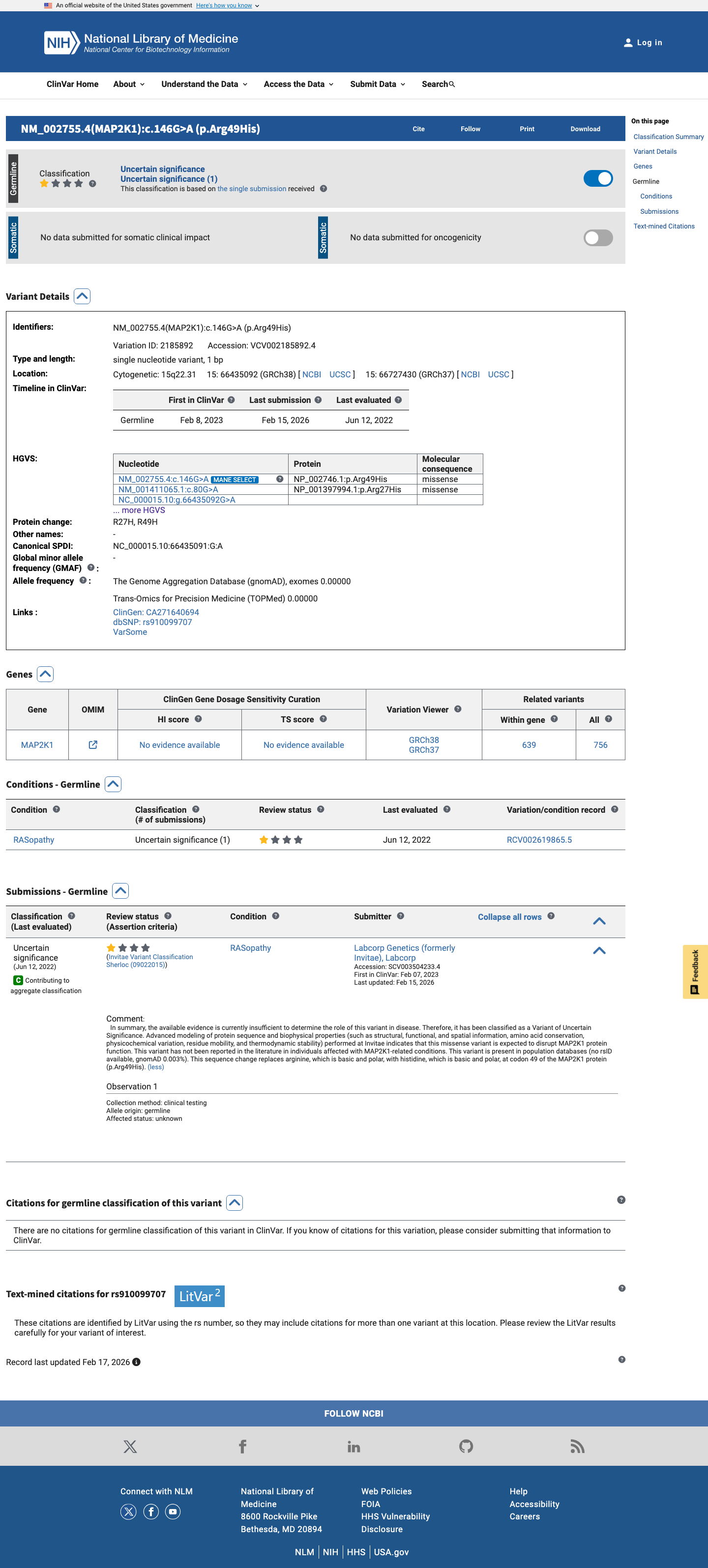
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 2185892)
In silico
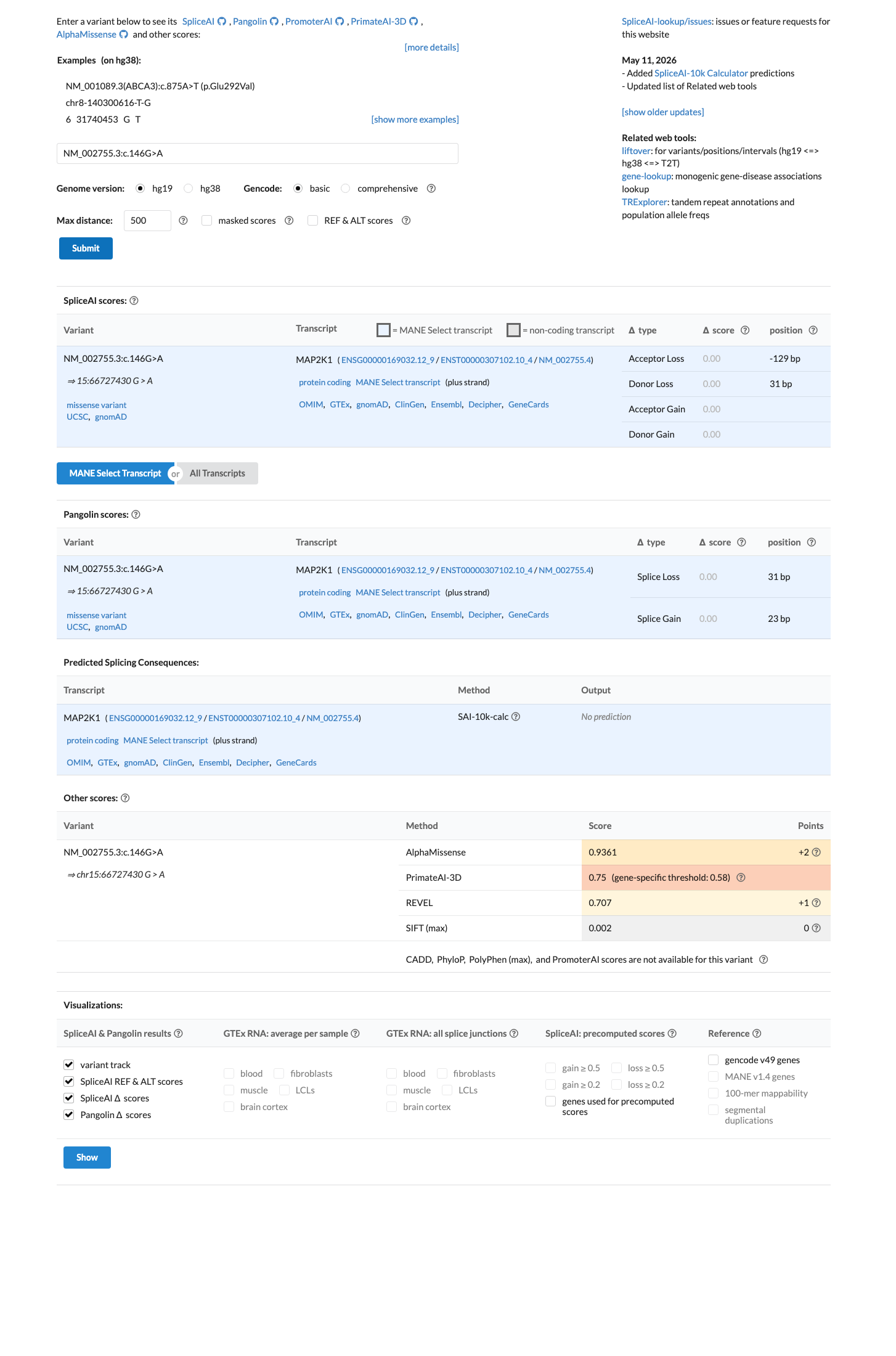
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.707. BayesDel score = 0.13946.
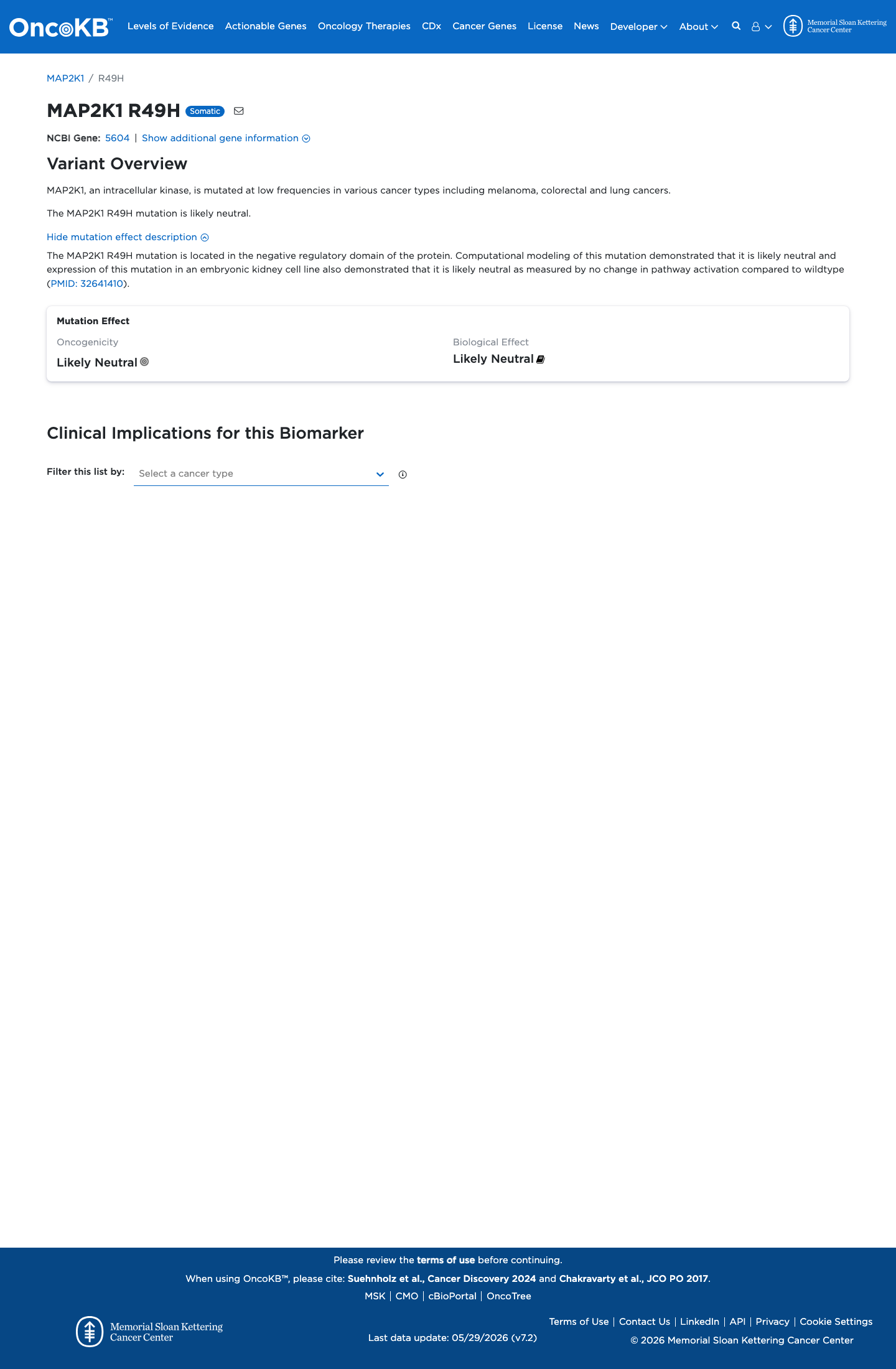
Functional
Likely Neutral
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Neutral; curated oncogenicity label: Likely Neutral.
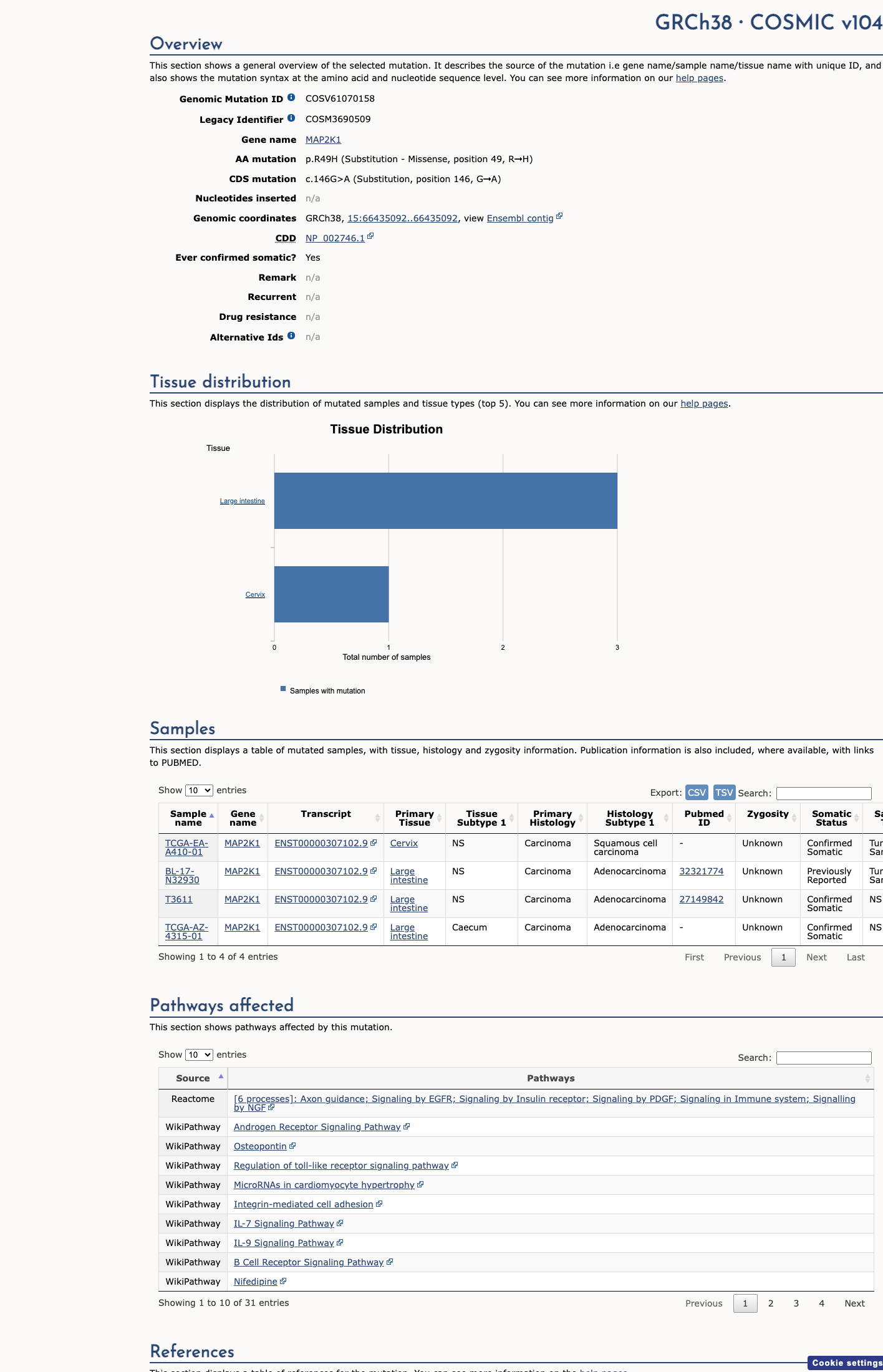
COSMIC
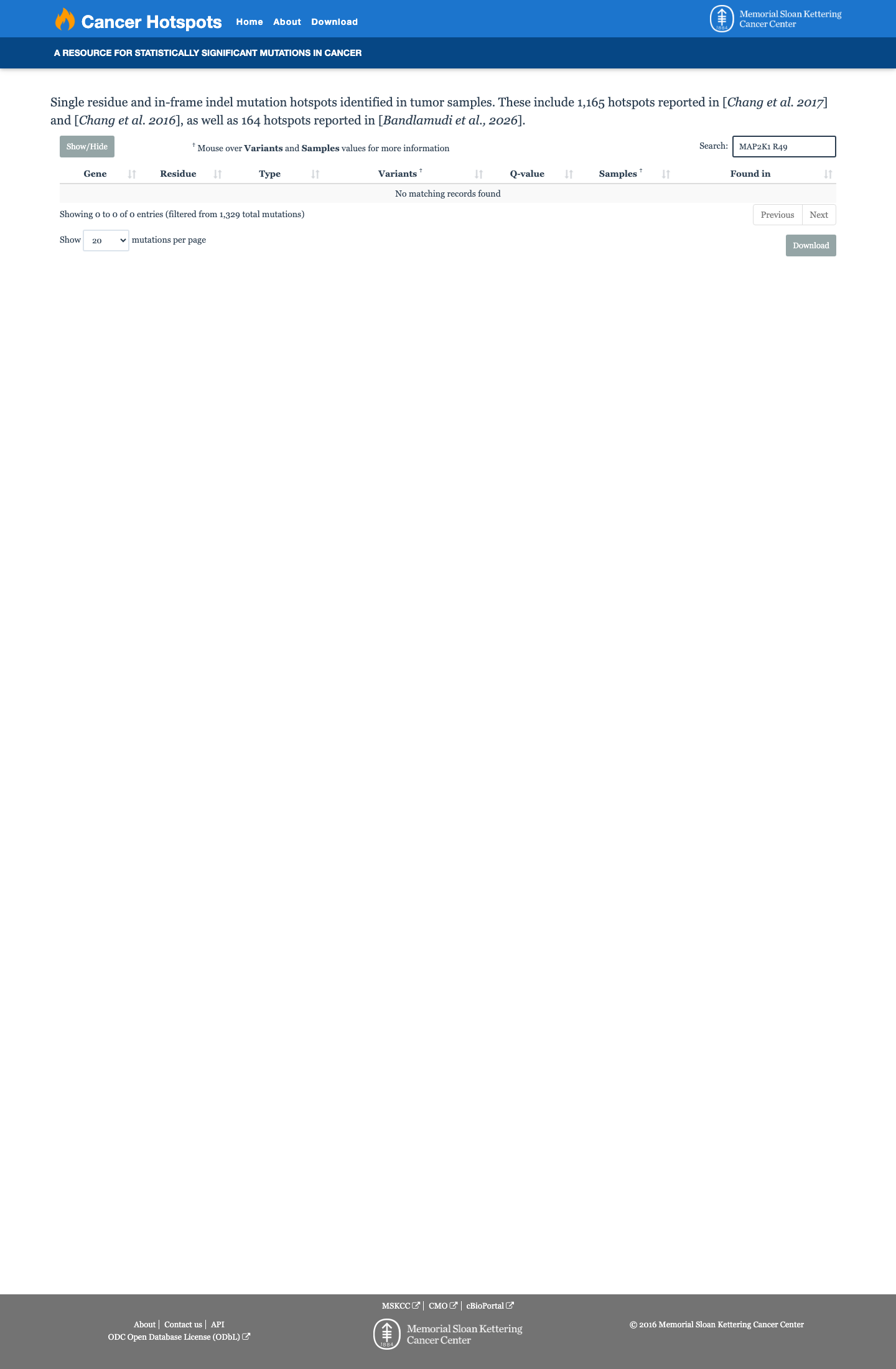
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV61070158, n = 4 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
32641410 ↗
Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer.
ONCOKB
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR