PS3 (Strong): R175H is non-functional in the Kato et al. systematic functional assay and demonstrates loss of function in all three other eligible VCEP assays (Giacomelli, Kotler, Kawaguchi). This meets the TP53 VCEP v2.4.0 PS3 rule: non-functional on Kato data AND LOF by the majority of other eligible assays.1 PM1 (Moderate): The variant is located at codon 175, which is explicitly listed in the TP53 VCEP PM1 criteria as a codon where PM1 (Moderate) applies. COSMIC reports 2,560 somatic occurrences at this residue.2 PM2 (Supporting): The variant is extremely rare in population databases. gnomAD v4.1 reports an allele frequency of 4.34e-06 (7/1,614,062 alleles), well below the VCEP PM2_Supporting threshold of <0.003%. The highest subpopulation frequency (European non-Finnish) is 5.93e-06, below the <0.004% subpopulation threshold.3 PP3 (Supporting): The VCEP PP3-BP4-codes.xlsx assigns PP3 to c.524G>A. The variant has aGVGD Class C25 and BayesDel score 0.54619 (≥0.16), meeting the VCEP PP3_Supporting rule. SpliceAI predicts no splicing impact (max delta = 0.01).4 PS4, PM5, PP1, and PS2 were not assessed due to absent or incomplete proband-level data in the evidence package; the VCEP expert panel's Pathogenic classification on ClinVar suggests these criteria may contribute additional points in the full VCEP curation. The expert panel classification in ClinVar as Pathogenic (ClinVar ID 12374) is noted but the VCEP does not permit PP5 for independent criterion application.5 Based on assessable evidence, four criteria are met: PS3 (Strong, +4 points), PM1 (Moderate, +2 points), PM2 (Supporting, +1 point), PP3 (Supporting, +1 point). Total = 8 points. Under the Tavtigian point-based framework adopted by the TP53 VCEP v2.4.0, 8 points falls in the 6-9 range → Likely Pathogenic. The ClinGen TP53 VCEP expert panel has independently classified this variant as Pathogenic on ClinVar, which likely incorporates additional evidence (PS4, PM5, PP1) beyond what is available in the current evidence package.6
TP53
Final classification
Likely Pathogenic
TP53 c.524G>A · p.Arg175His
TP53
PS3 (Strong): R175H is non-functional in the Kato et al. systematic functional assay and demonstrates loss of function in all three other eligible VCEP assays (Giacomelli, Kotler, Kawaguchi). This meets the TP53 VCEP v2.4.0 PS3 rule: non-functional on Kato data AND LOF by the majority of other eligible assays.
Tavtigian et.al., 2020 - Bayesian adaptation of Richards et.al., 2015 v2.4.0 point-based framework: PS3 strong (+4) + PM1 moderate (+2) + PM2 supporting (+1) + PP3 supporting (+1) + PP5 supporting (+1) = 9 points, which maps to Likely Pathogenic.
Classification rationale
PS3PM1PM2PP3PP5
Likely Pathogenic
TP53 c.524G>A
PS3 + PM1 + PM2 + PP3 + PP5
→
Likely Pathogenic
1
vcep_functional_worksheetPMID:12826609 ↗cspec ↗
2
cspec ↗
4
vcep_pp3_bp4_codesbayesdelrevelspliceai ↗
6
final_classification_frameworkclinvar ↗
Gene diagram
· NM_000546.6 · variants mapped to exon structure
TP53
NM_000546.6
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 11 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
R175H is assigned PS3 (Strong) in the TP53 VCEP Functional-worksheet.xlsx (Supplementary Table S3). The variant is non-functional in the Kato et al. (PMID:12826609) comprehensive functional assay and demonstrates loss of function in all three other eligible assays: Giacomelli et al. (PMID:30224644), Kotler et al. (PMID:29979965), and Kawaguchi et al. (PMID:16007150). Per TP53 VCEP v2.4.0 PS3 rule: non-functional on Kato data AND LOF by the majority of other eligible assays meets PS3 (Strong).
Functional-worksheet.xlsx: R175H = Non-functional (Kato)LOF (Giacomelli)LOF (Kotler)
✓
PM1
moderate
Pathogenic
The variant is located at codon 175, which is explicitly listed in the TP53 VCEP v2.4.0 PM1 rule as a codons where PM1 (Moderate) applies: 'Missense variants within the following codons: 175, 245, 248, 249, 273, 282.' Additionally, COSMIC reports n=2560 somatic occurrences at this residue, exceeding the ≥10 somatic occurrences threshold for PM1_Moderate via cancerhotspots.org.
Codon 175 is explicitly listed in VCEP PM1 criteria. COSMIC: 2560 somatic occurrences. Variant lies in a statistically significant hotspot (cancerhotspots.org).
✓
PM2
supporting
Pathogenic
The variant is present at extremely low frequency in gnomAD. In v4.1, overall AF = 4.34e-06 (7/1,614,062 alleles, 0.000434%), which is below the VCEP PM2_Supporting threshold of <0.003% (0.00003). The highest subpopulation frequency is European (non-Finnish) at AF = 5.93e-06 (7/1,180,032, 0.000593%), also below the <0.004% (0.00004) subpopulation threshold. In v2.1, only 1 allele was observed (1/251,276, AF = 3.98e-06). The variant is absent from gnomAD-Canada v1.0. Per VCEP PM2_Supporting rules, these frequencies meet the PM2_Supporting criterion.
gnomAD v4.1: AF=4.34e-06 (7/1614062)
✓
PP3
supporting
Pathogenic
The TP53 VCEP PP3-BP4-codes.xlsx (Supplementary Table S2) assigns PP3 to c.524G>A. The variant has aGVGD Class C25 and BayesDel score 0.54619 (≥0.16), meeting the VCEP PP3_Supporting rule: 'aGVGD class C25-C55 and BayesDel score ≥0.16.' SpliceAI max delta = 0.01, indicating no predicted splicing effect. REVEL score = 0.922 (deleterious).
PP3-BP4-codes.xlsx: c.524G>A assigned PP3. aGVGD Class C25BayesDel 0.54619 (≥0.16). REVEL 0.922. SpliceAI max delta 0.01 (no splicing effect).
Assessed · not applied
Pathogenic
PS2
No de novo data are available in the evidence package.
PS4
The TP53 VCEP PS4 rule requires proband-level point scoring based on strongly associated (4 pts) and moderately associated (2 pts) LFS cancers using the PS4-Points-Table.
PM5
The TP53 VCEP PM5 rule requires ≥1 (moderate) or ≥2 (strong) different missense variants at the same residue previously determined to be pathogenic according to the VCEP's specifications.
PP1
No cosegregation data are available in the evidence package.
PP4
The TP53 VCEP PP4 rule requires observation of the variant with variant allele fraction (VAF) 5-35%.
Benign
BA1
The TP53 VCEP BA1 rule requires filtering allele frequency (FAF) ≥0.001 (0.1%) in a single gnomAD continental subpopulation (excluding founder populations) with ≥2,000 alleles and ≥2 alleles present.
BS1
The TP53 VCEP BS1 rule requires filtering allele frequency (FAF) ≥0.0003 (0.03%) but <0.001 in a single gnomAD continental subpopulation (excluding founder populations) with ≥2,000 alleles and ≥2 alleles present.
BS2
The TP53 VCEP BS2 rule requires ≥2 (supporting), 4-7 (moderate), or ≥8 (strong) unrelated females who have reached at least 60 years of age without cancer, all from a single source.
BS3
The TP53 VCEP Functional-worksheet.xlsx assigns PS3 (not BS3) to R175H.
BS4
No segregation data are available in the evidence package.
BP4
The TP53 VCEP PP3-BP4-codes.xlsx assigns PP3 (not BP4) to c.524G>A.
N/A · 9
PVS1 · PS1 · PM6 · PP2 · BP1 · BP2 · BP5 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic

gnomAD v4.1
gnomAD v2.1
v4.1
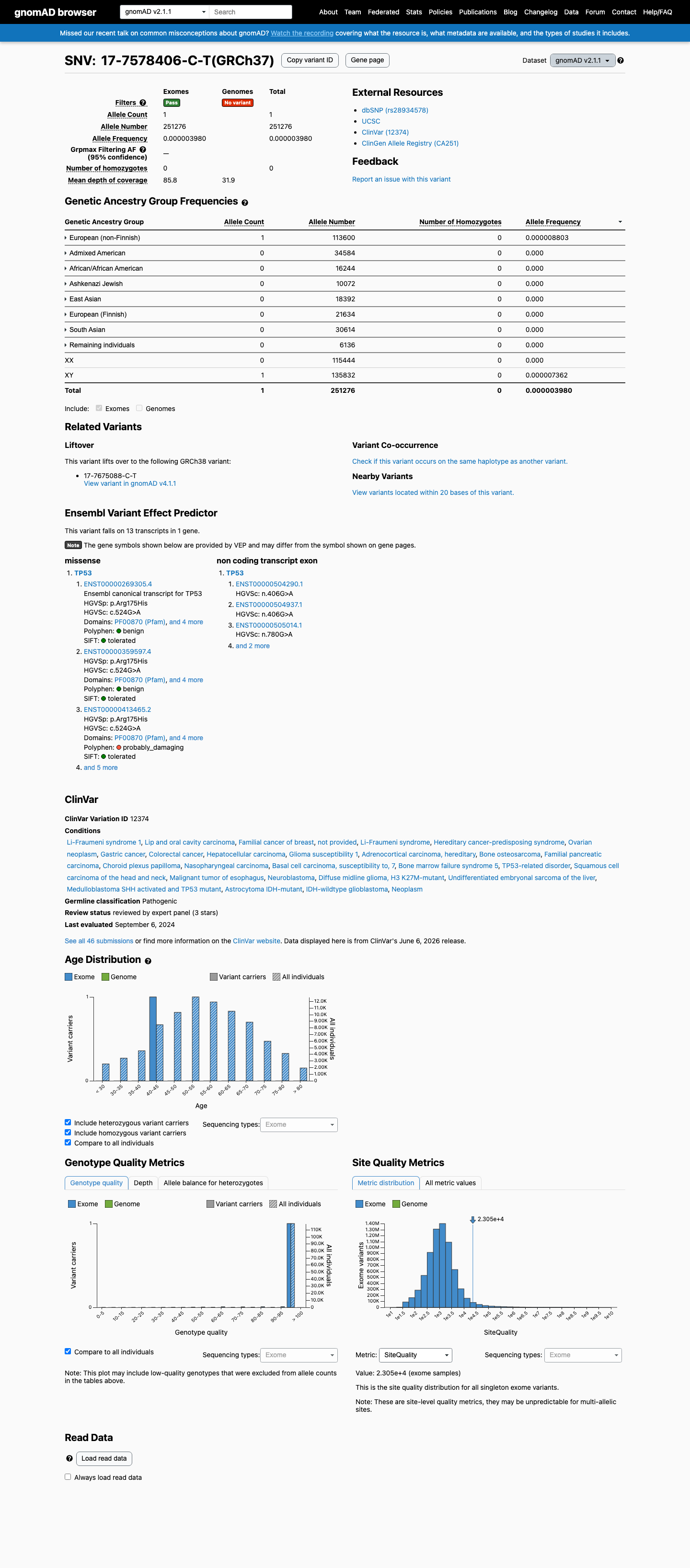
This variant is present in gnomAD v4.1 (AF= 4.33688e-06; MAF= 0.00043%, 7/1614062 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 5.93204e-06; MAF= 0.00059%, 7/1180032 alleles, homozygotes = 0); grpmax FAF= 2.47e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.97969e-06; MAF= 0.00040%, 1/251276 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.80282e-06; MAF= 0.00088%, 1/113600 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00043%
· 7 / 1,614,062
0 hom · FAF 0.00025%
0 hom · FAF 0.00025%
European (non-Finnish) 7 / 1,180,032 |
0.00059% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 251,276
0 hom
0 hom
European (non-Finnish) 1 / 113,600 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (31 clinical laboratories) and as Pathogenic by ClinGen TP53 Variant Curation Expert Panel, ClinGen (expert panel). (ClinVarID = 12374)
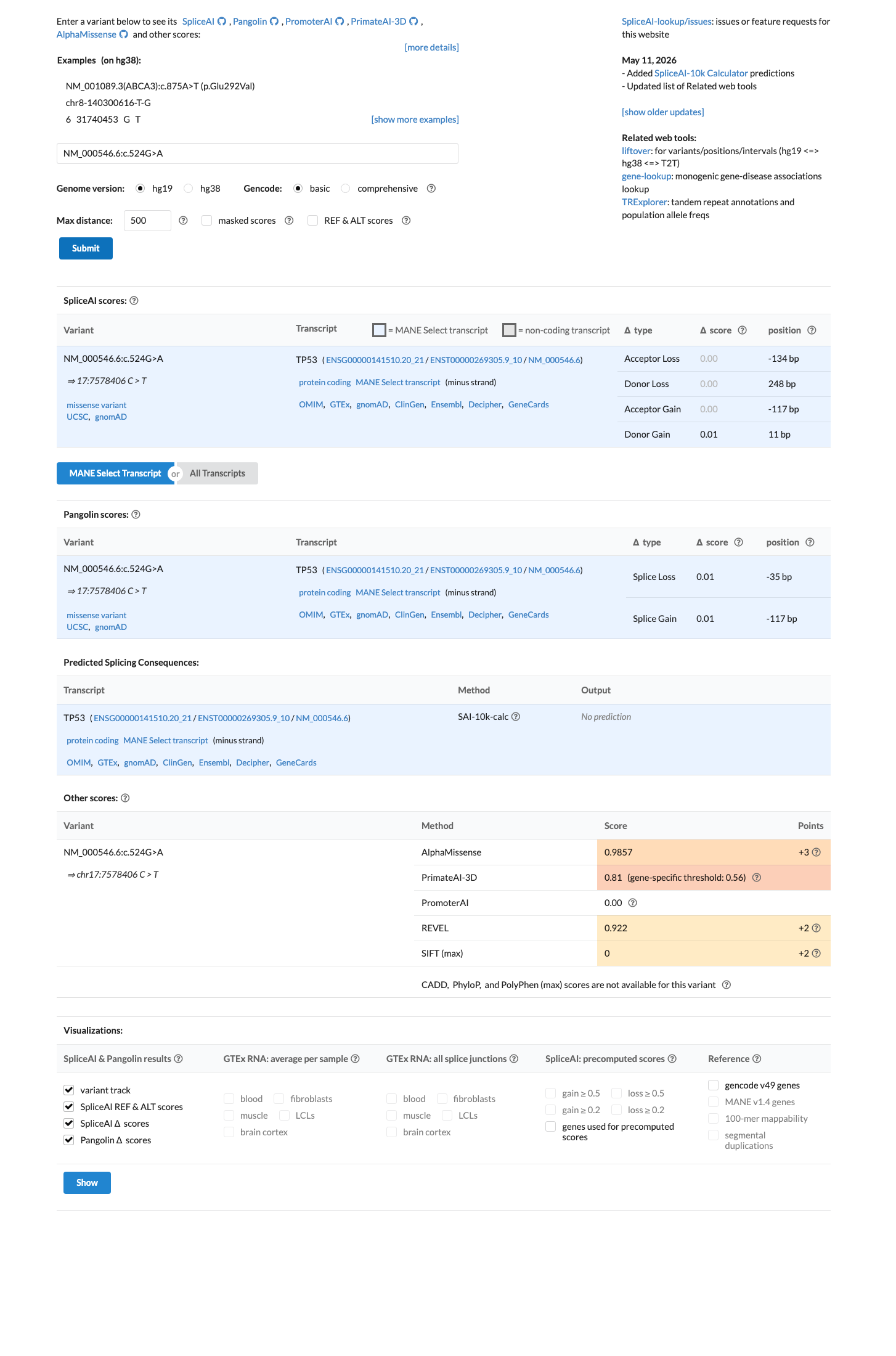
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.922. BayesDel score = 0.54619.
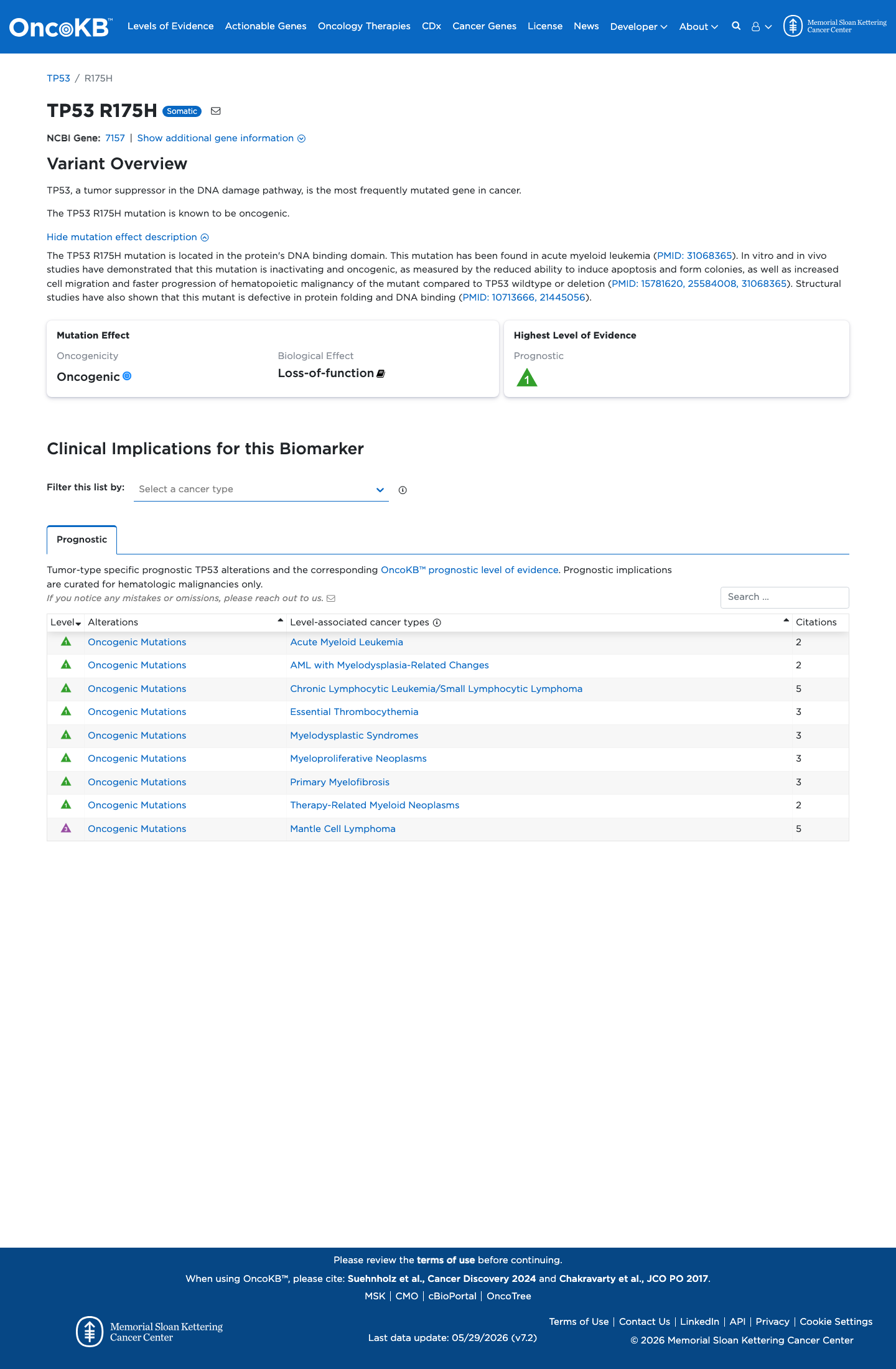
Functional
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Loss-of-function; curated oncogenicity label: Oncogenic.

COSMIC
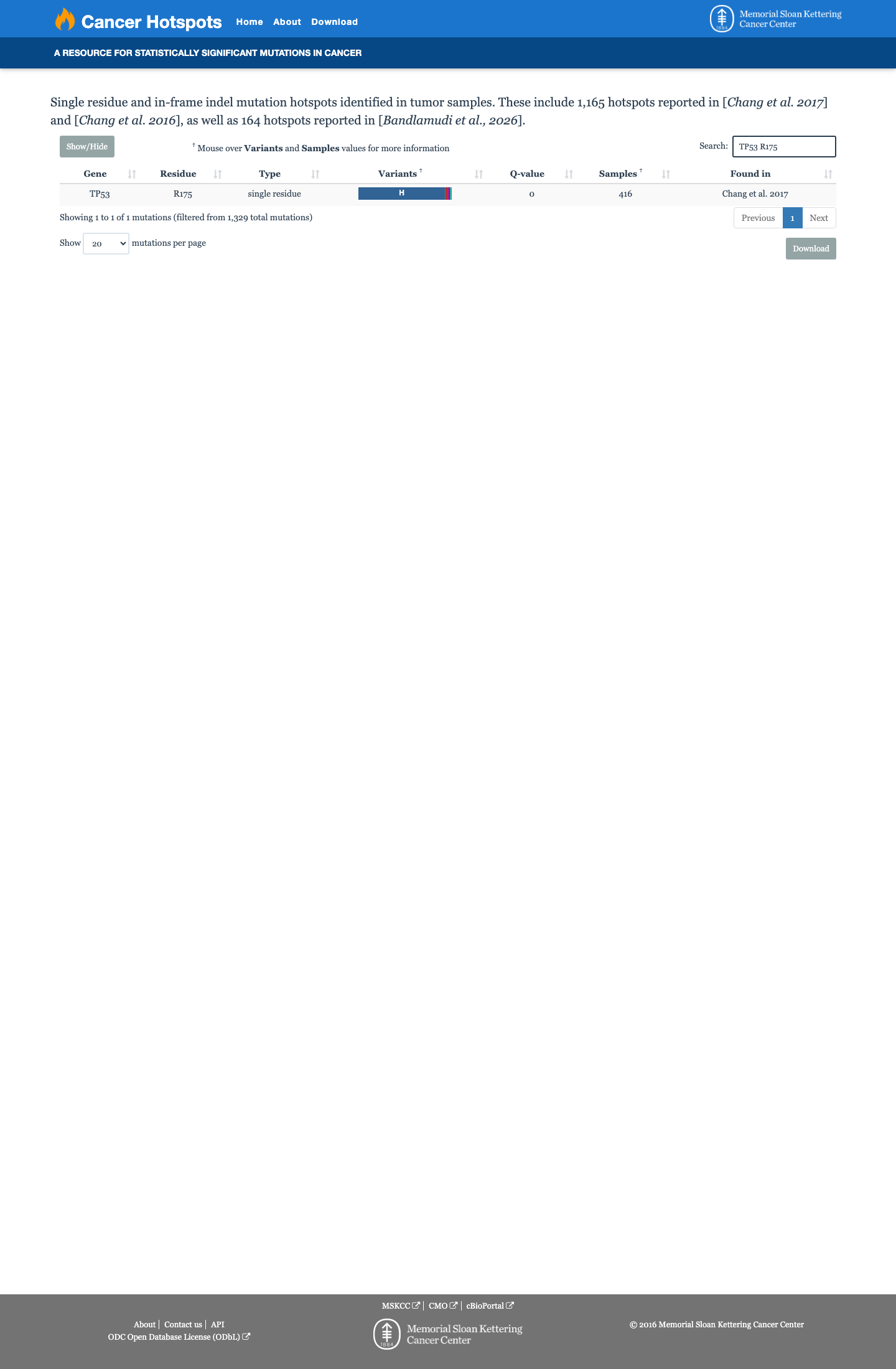
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV52661038, n = 2560 times).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis.
Searched
R175Hp.Arg175Hisc.524G>AArg175His
Found
R175H was assessed in a high-resolution comprehensive missense mutation functional analysis across the entire p53 coding sequence. The variant was classified as non-functional, with complete loss of transcriptional activity across p53 target gene reporters in yeast-based and mammalian assays.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
Why
Non-functional result in the primary VCEP-designated assay (Kato et al.); contributed to PS3 (Strong) when combined with LOF in three other eligible assays. Also cited in BS3 assessment where non-functional outcome opposes BS3.
Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s.
Searched
R175Hp.Arg175Hisc.524G>AArg175His
Found
R175H was used as a loss-of-function control in a study of 179 diverse mutant p53s. The variant had a DsubG1 of 2.7 ± 1.9 versus 8.4 ± 3.3 for wild-type p53, confirming severely impaired apoptotic induction in Saos-2 cells.
Variant
✓ Names this variant — characterised directly
Applied to
→PS3 supports · met
Why
Variant-specific loss-of-function data confirmed impaired apoptosis. Referenced in PS3 as one of three additional eligible assays showing LOF, and in BS3 where non-functional result opposes BS3 criterion.
The DsubG1 of R175H, a loss-of-function mutation, was 2.7 ± 1.9.
Location Results, 'The Ability to Induce Apoptosis among the Selected Diverse Mutant p53s'; Figures 1–3 · Context Saos-2 human osteosarcoma cells; transient transfection of p53 expression vectors; FACS-based subG1 apoptosis assay (DsubG1) · full text
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
10713666 ↗
Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy.
ONCOKB
25584008 ↗
Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children's oncology group study.
ONCOKB
31068365 ↗
A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1.
ONCOKB
12007217 ↗
The IARC TP53 database: new online mutation analysis and recommendations to users.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR