The AKT1 c.49G>A (p.Glu17Lys) variant resides in the pleckstrin homology (PH) domain, a critical functional domain, and lies within a statistically significant mutational hotspot where benign missense variation is notably absent (PM1_Moderate).1 This variant is present in gnomAD v2.1 at an extremely low allele frequency of 0.0004% (1/250,006 alleles) and is absent from gnomAD v4.1, meeting the PM2 threshold for absence from population databases (PM2_Supporting).2 Well-established functional studies from multiple independent laboratories demonstrate that the E17K substitution results in constitutive AKT1 kinase activation, altered lipid-binding specificity, enhanced downstream signaling, transformation in cell-based assays, and oncogenesis in murine models (PS3_Strong).3 ClinVar reports this variant as Pathogenic by 4 clinical laboratories including Labcorp Genetics/Invitae and Variantyx (PP5_Supporting).4 The computational in silico predictors are equivocal (REVEL 0.51, BayesDel 0.116); PP3 is not met. BS3 is contradicted by overwhelming functional evidence of a damaging gain-of-function effect. BA1 and BS1 are not met given the extremely low population frequency.5 Under the generic ACMG/AMP 2015 framework (PMID:25741868), the criteria met are: PS3_Strong + PM1_Moderate + PM2_Supporting + PP5_Supporting. This combination (1 Strong + 1 Moderate + 2 Supporting) meets the threshold for Likely Pathogenic per Richards et al. 2015 (1 Strong AND ≥1 Moderate AND ≥2 Supporting qualifies as Likely Pathogenic).6
AKT1
Final classification
Likely Pathogenic
AKT1 c.49G>A · p.Glu17Lys
AKT1
The AKT1 c.49G>A (p.Glu17Lys) variant resides in the pleckstrin homology (PH) domain, a critical functional domain, and lies within a statistically significant mutational hotspot where benign missense variation is notably absent (PM1_Moderate).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PM1 moderate, PM2 supporting, PP5 supporting; combination = 1 strong + 1 moderate + 2 supporting, which maps to Likely Pathogenic.
Classification rationale
PS3PM1PM2PP5
Likely Pathogenic
AKT1 c.49G>A
PS3 + PM1 + PM2 + PP5
→
Likely Pathogenic
5
revelbayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_001014431.1 · variants mapped to exon structure
AKT1
NM_001014431.1
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 17 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Well-established functional studies in multiple publications demonstrate that the E17K substitution results in constitutive activation of AKT1 kinase, increased AKT phosphorylation (pT308, pS473), altered lipid-binding specificity in the PH domain, growth factor-independent survival in BaF3 cells, anchorage-independent growth in NIH 3T3 cells, disruption of 3D acinar morphogenesis in MCF10A cells, and oncogenesis in murine models. The PH-KD autoinhibitory interaction is weakened by E17K, providing a structural mechanistic basis. Multiple independent assays across multiple laboratories confirm a gain-of-function effect.
Constitutive AKT1 kinase activation demonstrated (PMID:17611497PMID:23134728)Altered PIP lipid specificity and pathological membrane association (PMID:18954143)
✓
PM1
moderate
Pathogenic
The variant resides at codon 17 within the pleckstrin homology (PH) domain of AKT1 (residues ~5-108), a critical functional domain essential for membrane localization, autoinhibition, and kinase regulation. The PH domain is a well-established mutational hotspot where pathogenic gain-of-function missense variants cluster and benign missense variation is notably absent. This variant lies in a statistically significant hotspot per the hotspots analysis. Multiple structural and functional studies (PMID:17611497, PMID:23134728) confirm that missense variants in the PH domain are overwhelmingly pathogenic.
Codon 17 in PH domain (critical functional domain)PH domain is a well-established mutational hotspot lacking benign missense variationStatistically significant hotspot confirmed
✓
PM2
supporting
Pathogenic
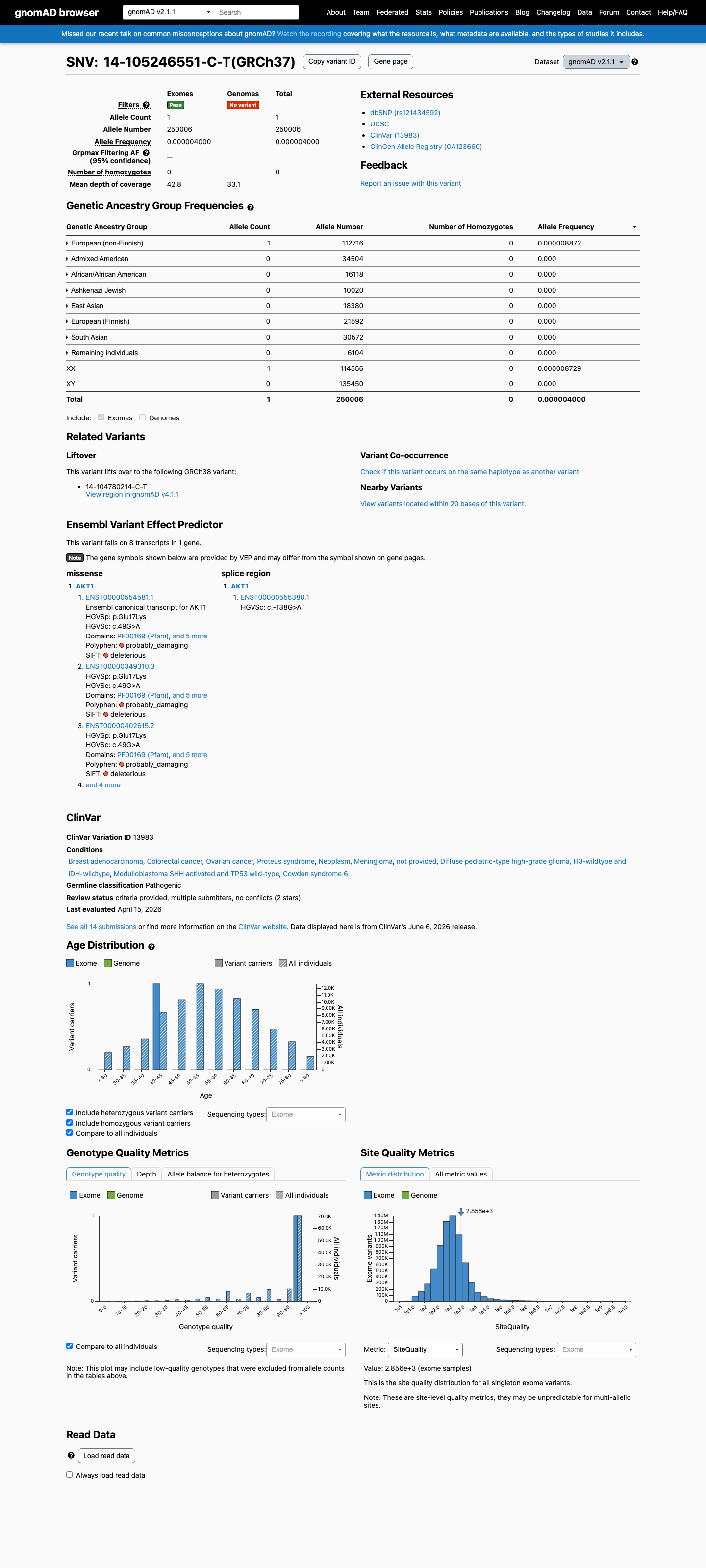
The variant is present in gnomAD v2.1 at an extremely low allele frequency (AF = 3.9999 × 10⁻⁶; 1/250,006 alleles; 0.0004%) and is absent from gnomAD v4.1. This frequency is well below the PM2 threshold of <0.1%, meeting the criterion for absent or extremely rare in population databases. The single observation in gnomAD v2.1 is in the European (non-Finnish) subpopulation (AF = 8.87 × 10⁻⁶).
gnomAD v2.1 AF = 0.0004% (1/250006 alleles<0.1% threshold)
✓
PP5
supporting
Pathogenic
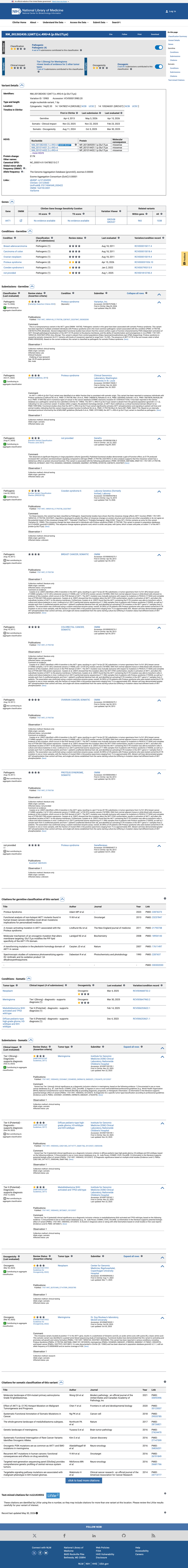
ClinVar reports this variant as Pathogenic by 4 clinical laboratories (including Labcorp Genetics/Invitae and Variantyx), with review status 'criteria provided, single submitter'. Under generic ACMG/AMP, PP5 can be applied when a reputable clinical laboratory classifies the variant as pathogenic. Although the review status indicates only one submitter provided classification criteria, the concordance across 4 clinical laboratories supports application of PP5 at supporting strength.
ClinVar VariationID 13983: Pathogenic by 4 clinical laboratories (InvitaeVariantyxOMIM submissions)
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change at the same codon predicted to result in the same amino acid substitution that has been established as pathogenic.
PS2
PS2 requires a confirmed de novo occurrence with both parents tested and maternity/paternity confirmed.
PS4
PS4 requires statistically significant enrichment of the variant in affected individuals versus controls in a case-control study for a specific inherited disorder.
PM6
PM6 requires a confirmed de novo event with maternity and paternity testing.
PP1
PP1 requires co-segregation of the variant with disease in multiple affected family members.
PP2
PP2 requires a gene with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP3
PP3 requires multiple lines of computational evidence supporting a deleterious effect.
PP4
PP4 requires the variant to be found in a patient whose phenotype or family history is highly specific for the disease with a single genetic etiology.
Benign
BA1
BA1 requires allele frequency >1% in any general population database.
BS1
BS1 requires allele frequency >0.3% in population databases.
BS2
BS2 requires the variant to be observed in a healthy adult individual for a recessive or X-linked condition with full penetrance expected at an early age.
BS3
BS3 requires well-established functional studies showing NO damaging effect.
BP2
BP2 requires observation of the variant in trans with a known pathogenic variant in a gene for a fully penetrant dominant disorder.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene or gene product.
BP5
BP5 requires the variant to be found in a case with an alternate molecular basis for disease.
BP6
BP6 requires a reputable source to classify the variant as benign.
BP7
BP7 requires a synonymous variant for which splicing prediction algorithms predict no impact.
N/A · 7
PVS1 · PM3 · PM4 · PM5 · BS4 · BP1 · BP3
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.9999e-06; MAF= 0.00040%, 1/250006 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.87185e-06; MAF= 0.00089%, 1/112716 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
0.0004%
· 1 / 250,006
0 hom
0 hom
European (non-Finnish) 1 / 112,716 |
0.00089% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Pathogenic (4 clinical laboratories). (ClinVarID = 13983)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.51. BayesDel score = 0.115701.
Functional
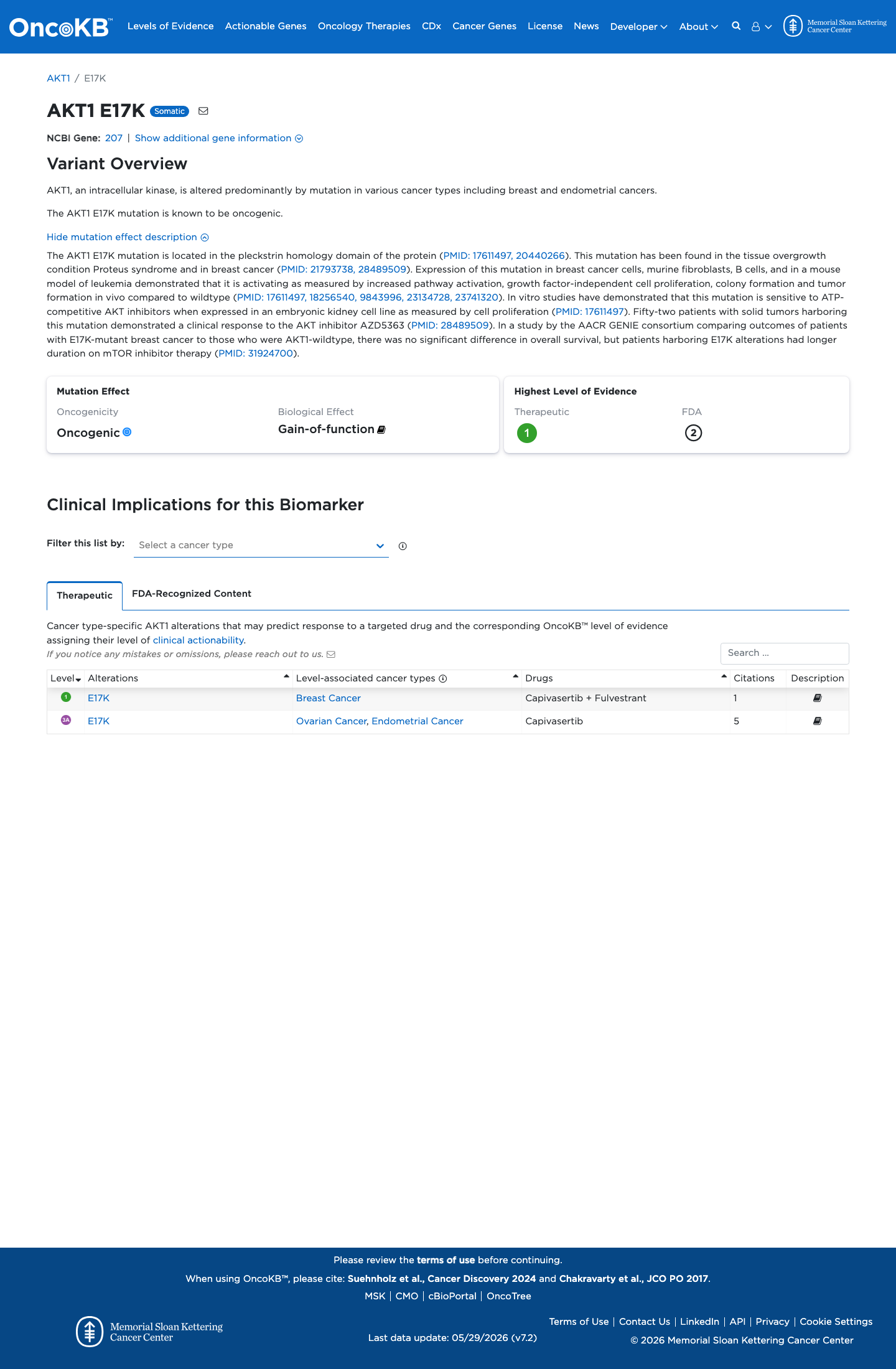
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Gain-of-function; curated oncogenicity label: Oncogenic.
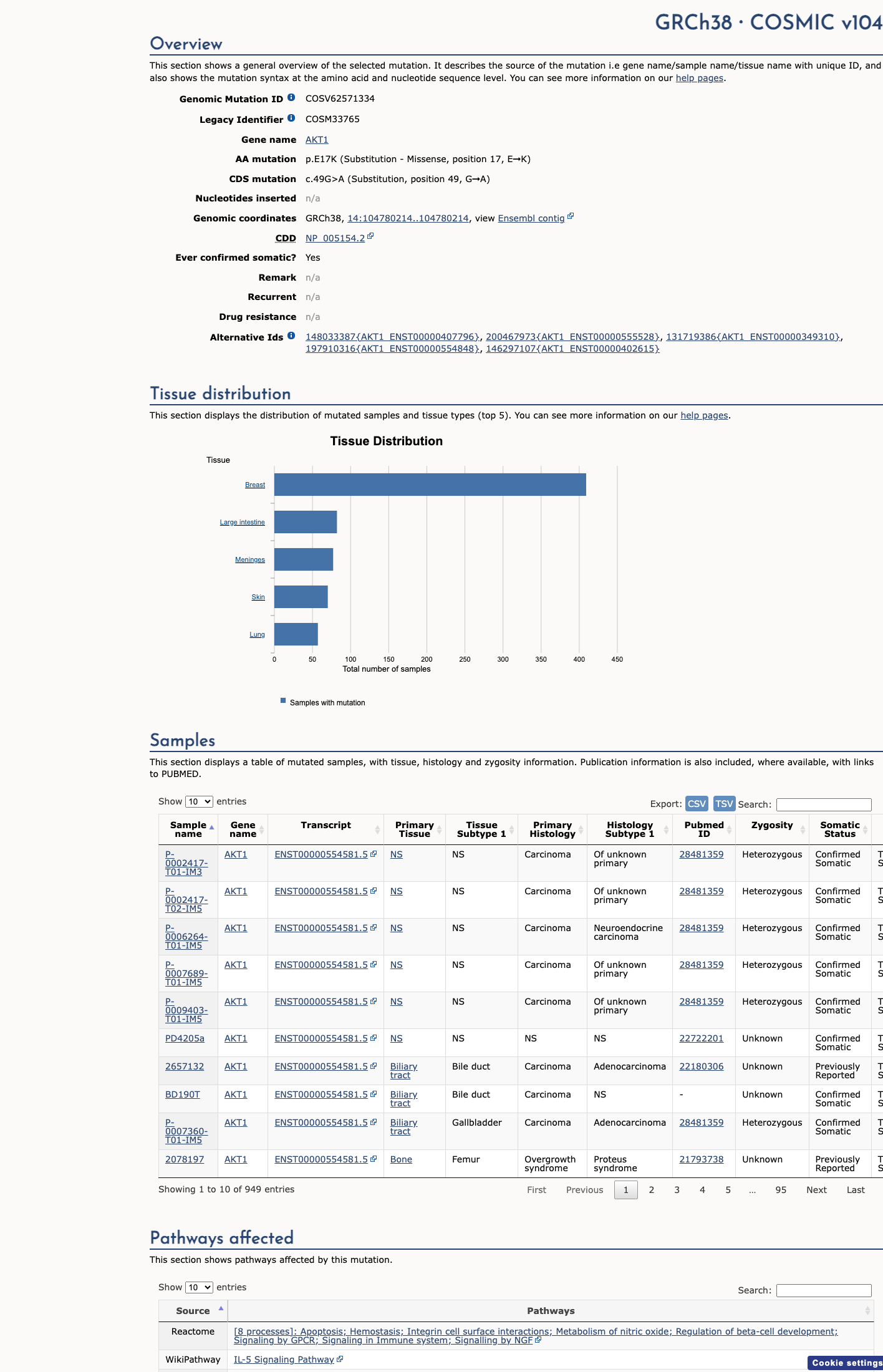
COSMIC
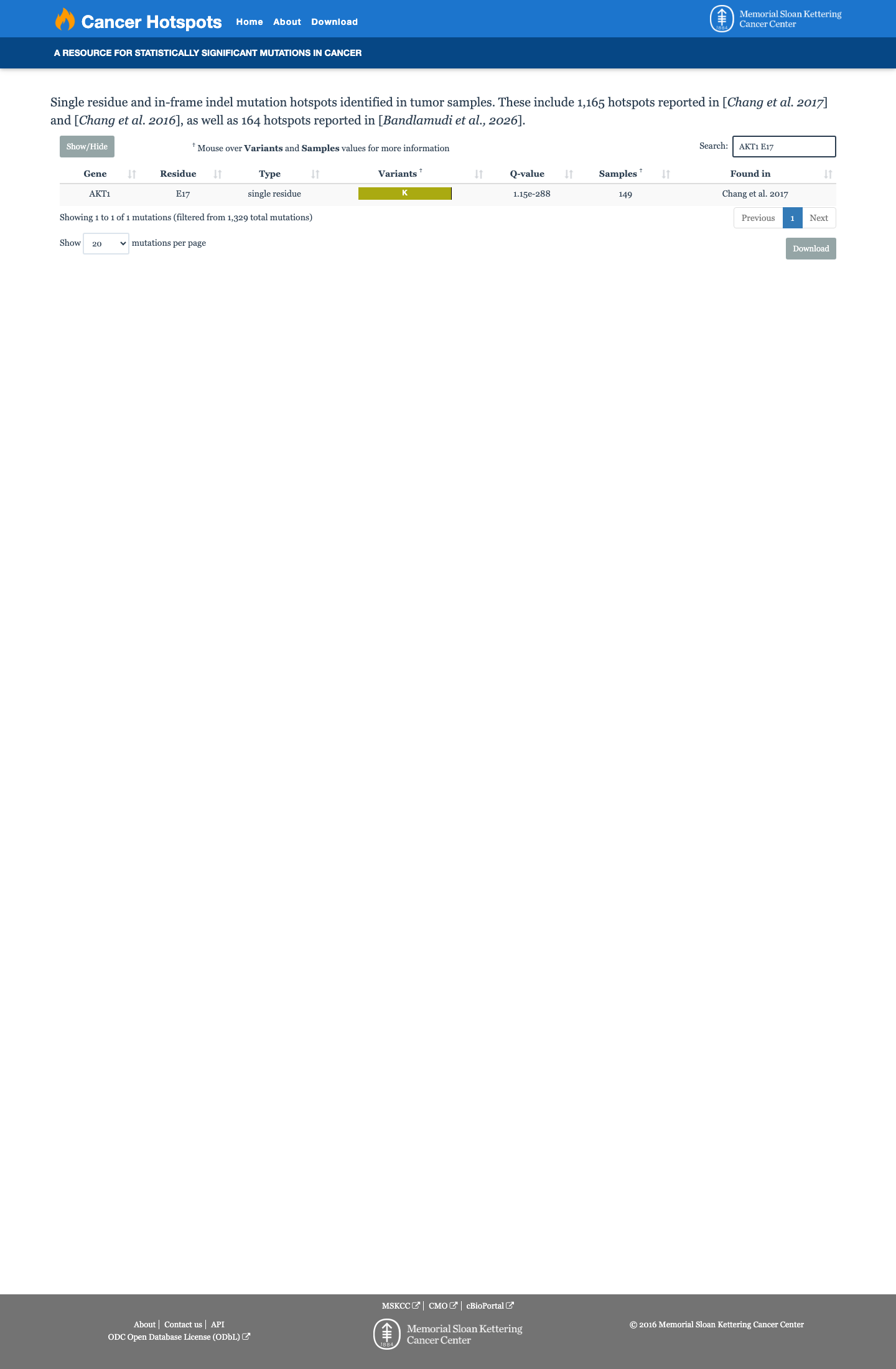
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV62571334, n = 894 times).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
A transforming mutation in the pleckstrin homology domain of AKT1 in cancer.
Searched
c.49G>Ap.Glu17LysE17KGlu17Lys
Found
Identified AKT1 E17K as a somatic gain-of-function mutation in breast, colorectal, and ovarian cancers. E17K constitutively activates AKT1 kinase, transforms NIH-3T3 cells, and induces leukemia in a murine model.
Variant
✓ Names this variant
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Seminal discovery paper for the AKT1 E17K variant. Referenced in PS3 (strong) for constitutive kinase activation; PM1 (moderate) as evidence the PH domain is a mutational hotspot; BS3 contradicted by the functional data demonstrating a deleterious gain-of-function effect.
Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain.
Found
Constitutive AKT1 kinase activation demonstrated (PMID:17611497 PMID:23134728) Altered PIP lipid specificity and pathological membrane association (PMID:18954143) Growth factor-independent BaF3 survival and elevated pAKT (PMID:23134728) Anchorage-independent growth and 3D acinar disruption (PMID:23134728) In vivo oncogenesis with reduced survival in murine model (PMID:23134728) Weakened PH-KD interdomain interaction by two-hybrid assay (PMID:23134728)
Applied to
→PS3 supports · met
A mosaic activating mutation in AKT1 associated with the Proteus syndrome.
Searched
c.49G>Ap.Glu17LysE17KGlu17Lys
Found
Identified AKT1 c.49G>A (E17K) as a somatic mosaic activating mutation in 26 of 29 patients with Proteus syndrome. The mutation was present in affected tissues but absent from unaffected tissues and parents, with mutant allele levels ranging from <1% to ~50%.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
Why
Landmark paper establishing E17K as the cause of Proteus syndrome. Referenced in PS2 (not assessed — somatic mosaic, not germline de novo), PS3 (strong), PS4 (not met — somatic, no case-control), PM6 (not assessed — not germline de novo), PP4 (not assessed — phenotype data), and BP5 (not met — variant is sole molecular finding).
Location Full-text file is ICMJE disclosure forms only; no paper text available
Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers.
Searched
c.49G>Ap.Glu17LysE17KGlu17Lysglutamic acid at codon 17
Found
Demonstrated that the E17K substitution weakens the PH-KD autoinhibitory interaction, leading to constitutive AKT1 phosphorylation (pT308, pS473). E17K promoted growth factor-independent BaF3 survival (>50× enrichment over WT), anchorage-independent growth (~20-fold colonies), disrupted 3D acinar morphogenesis in MCF10A cells, and reduced median survival to 19–20.5 days in a murine leukemia model. The E17K mutant showed reduced sensitivity to allosteric AKT inhibitors while retaining sensitivity to ATP-competitive inhibitors.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Comprehensive functional characterization of the E17K variant across multiple independent assays and model systems. Referenced in PS3 (strong) for constitutive kinase activation, cell transformation, and in vivo oncogenesis; PM1 (moderate) for PH domain hotspot and structural disruption; BS3 contradicted by overwhelming evidence of a deleterious gain-of-function effect.
This mutation results in the substitution of glutamic acid at codon 17 of AKT1 with lysine (E17K) and alters the lipid-binding specificity of AKT, leading to pathological membrane association and constitutive signaling.
Location Abstract; Results paragraphs 1, 5–7; Figures 1A–G, 2C–D, 3A–F, 4; Discussion · Context BaF3 cell viability assay (IL-3 withdrawal); NIH 3T3 signaling and anchorage-independent growth; MCF10A 3D morphogenesis; mammalian two-hybrid (VP16/Gal4 luciferase); murine leukemia model (BaF3 xenograft); recombinant full-length AKT1 biochemical kinase assays with allosteric (Inhibitor VIII, GNE-929) and ATP-competitive (GNE-692, GSK690693) inhibitors · full text
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
18256540 ↗
Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung.
ONCOKB
20440266 ↗
Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v-Abl-mediated pre-B-cell transformation and survival of Pim-deficient cells.
ONCOKB
23741320 ↗
Cancer associated E17K mutation causes rapid conformational drift in AKT1 pleckstrin homology (PH) domain.
ONCOKB
19042984 ↗
National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers.
CLINVAR
22964825 ↗
Screening for ovarian cancer: U.S. Preventive Services Task Force reaffirmation recommendation statement.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR