NM_004380.2:c.3609+1G>T is a canonical splice donor (+1) variant in CREBBP. Loss of function in CREBBP is an established mechanism for Rubinstein-Taybi syndrome (PMID:41153422). Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical splice variants in genes with established LOF disease mechanism are assigned PVS1 at very strong strength.1 The variant is absent from gnomAD v2.1 and v4.1 population databases (0 alleles observed), meeting PM2 at supporting strength under the generic ACMG/AMP <0.1% allele frequency threshold.2 SpliceAI predicts strong disruption of the canonical splice donor (max delta score = 1.00; donor loss = 1.0, donor gain = 0.84). This in silico evidence is consistent with PVS1 but is not separately scored as PP3 per PMC6185798 guidance to avoid double-counting splice prediction evidence.3 No verified clinical observations (de novo status, case counts, co-segregation, or functional data) for this exact variant were identified. ClinVar has no entry. Multiple criteria (PS2, PS3, PS4, PM6, PP1, PP4) could not be assessed due to absence of variant-specific evidence.4 Under the generic ACMG/AMP 2015 final classification rules (PMID:25741868), PVS1 (very strong) with PM2 (supporting) does not meet the threshold for Pathogenic (requires ≥2 supporting or 1 moderate + 1 supporting with PVS1) or Likely Pathogenic (requires PVS1 + 1 moderate). The variant is classified as a Variant of Uncertain Significance (VUS). Clinical corroboration through case-level evidence (de novo observation, co-segregation, or functional studies) would be needed to resolve the classification.5
CREBBP
Final classification
VUS
CREBBP c.3609+1G>T · p.?
CREBBP
NM_004380.2:c.3609+1G>T is a canonical splice donor (+1) variant in CREBBP. Loss of function in CREBBP is an established mechanism for Rubinstein-Taybi syndrome (PMID:41153422). Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical splice variants in genes with established LOF disease mechanism are assigned PVS1 at very strong strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 supporting; combination = 1 very strong + 1 supporting, which maps to VUS.
Classification rationale
PVS1PM2
VUS
CREBBP c.3609+1G>T
PVS1 + PM2
→
VUS
5
generic_acmg_combination_rules
Gene diagram
· NM_004380.2 · variants mapped to exon structure
CREBBP
NM_004380.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 16 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_004380.2:c.3609+1G>T is a canonical splice donor (+1) variant in CREBBP, a gene for which loss of function is an established mechanism of Rubinstein-Taybi syndrome (RSTS). Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical splice variants in genes with established LOF disease mechanism receive PVS1 at very strong strength. The affected intron 18 (of 31 exons) is not in a biologically irrelevant distal region and NMD is predicted.
Canonical +1 splice donor disruption of CREBBPCREBBP loss of function is a well-established mechanism for Rubinstein-Taybi syndromePMC6185798 canonical splice PVS1 guidance applied
✓
PM2
supporting
Pathogenic
NM_004380.2:c.3609+1G>T is absent from gnomAD v2.1 and v4.1 population databases, meeting the PM2 allele frequency threshold (<0.1%) under generic ACMG/AMP 2015 rules.
Absent from gnomAD v2.1 (exomes0 alleles observed)Absent from gnomAD v4.1 (exomes
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant at the same nucleotide position with a different nucleotide change has been identified in ClinVar or the literature.
PS2
De novo observation data could not be verified.
PS3
No functional assay data specific to NM_004380.2:c.3609+1G>T were identified.
PS4
No verified case counts or case-control data for NM_004380.2:c.3609+1G>T were found.
PM1
This variant is not located in a recognized mutational hotspot or critical functional domain without benign variation.
PM6
Assumed de novo data could not be verified.
PP1
No verified co-segregation data for NM_004380.2:c.3609+1G>T in affected families were found.
PP3
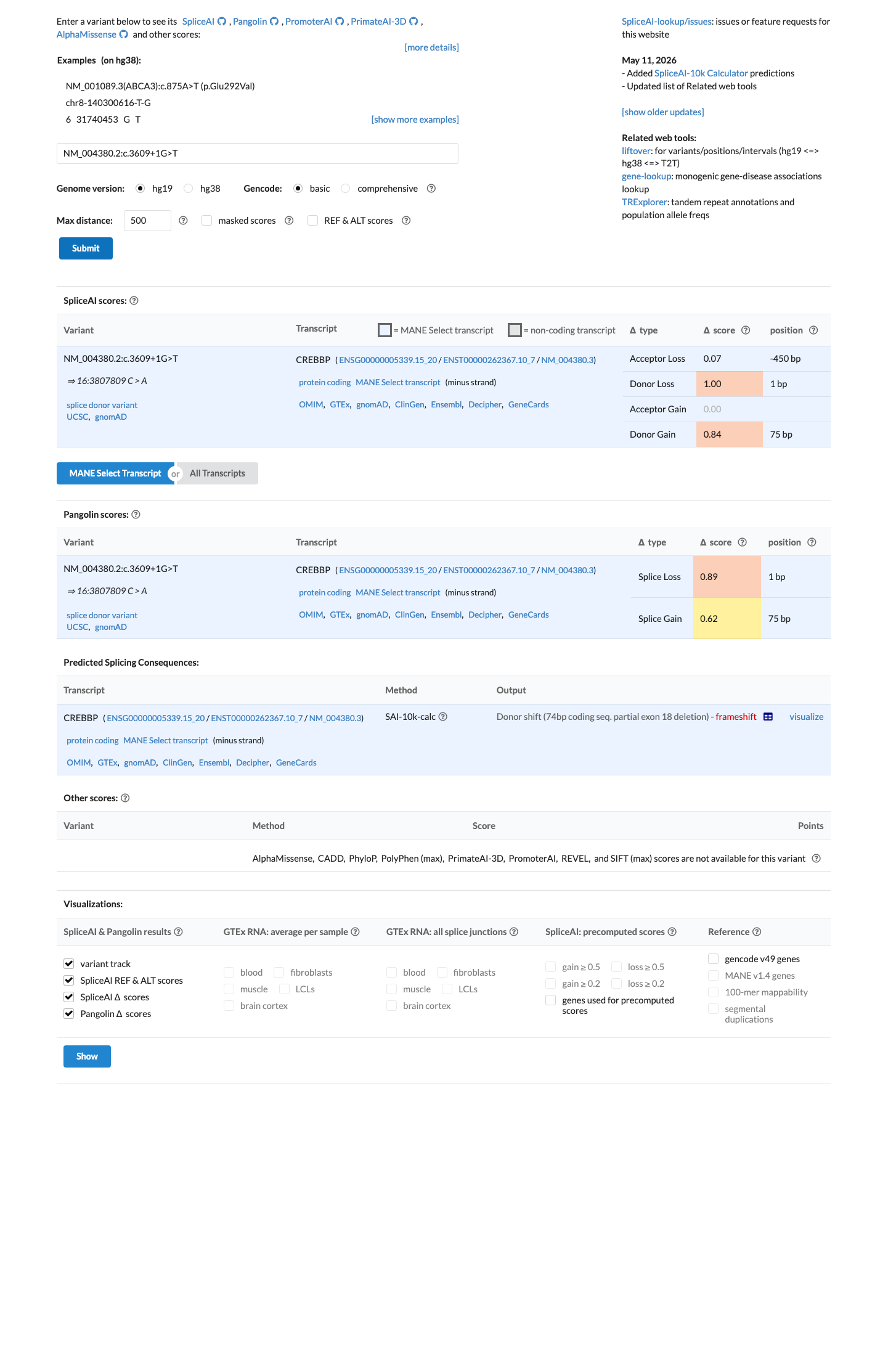
Multiple in silico tools support a deleterious effect on splicing (SpliceAI max delta = 1.0, donor loss = 1.0, donor gain = 0.84), and BayesDel score is 0.66.
PP4
No detailed phenotype information for patients carrying NM_004380.2:c.3609+1G>T was available to assess specificity for Rubinstein-Taybi syndrome.
PP5
No reputable source has reported NM_004380.2:c.3609+1G>T as pathogenic.
Benign
BA1
NM_004380.2:c.3609+1G>T is absent from gnomAD v2.1 and v4.1.
BS1
NM_004380.2:c.3609+1G>T is absent from gnomAD v2.1 and v4.1.
BS2
BS2 requires observation of the variant in a healthy adult individual in a pattern inconsistent with disease penetrance (e.g., homozygous in a dominant disorder).
BS3
No functional studies demonstrating a benign effect of NM_004380.2:c.3609+1G>T were identified.
BS4
BS4 requires lack of segregation in an affected family member.
BP4
Multiple in silico tools predict a deleterious effect: SpliceAI max delta = 1.0 (donor loss = 1.0, donor gain = 0.84) and BayesDel = 0.66 (above typical benign thresholds).
N/A · 10
PM3 · PM4 · PM5 · PP2 · BP1 · BP2 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 1.00). BayesDel score = 0.66.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
PMID 41153422
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
Sources & reference links