BA1 is met: allele frequency of 1.38–1.40% in the European (Finnish) population in gnomAD (v2.1: 347/25116 alleles, 3 homozygotes; v4.1: 896/64030 alleles, 11 homozygotes), exceeding the 1% stand-alone benign threshold.1 BS1 is met: global allele frequency of 0.36–0.39% in gnomAD (v2.1: 1093/282512 alleles; v4.1: 5806/1,612,416 alleles), exceeding the 0.3% strong benign threshold for a fully penetrant autosomal dominant disorder.2 BS2 is met: this variant has been observed in the homozygous state in 7 individuals in gnomAD v2.1 and 28 individuals in gnomAD v4.1. For a highly penetrant autosomal dominant tumor suppressor syndrome, biallelic inactivation would be expected to cause severe or lethal disease; observation of homozygotes in a general population database constitutes strong evidence for a benign role.3 BP4 is met: multiple lines of computational evidence (REVEL=0.338, BayesDel=−0.064, SpliceAI max delta=0.09) predict no damaging effect on the gene product.4 BP6 is met: ClinVar reports this variant as Benign by 23 clinical laboratories and Likely benign by 4 clinical laboratories (Variation ID: 5103), representing a strong consensus among diagnostic laboratories for a benign classification.5 PVS1 is not applicable: this is a missense variant (p.His732Tyr) and does not meet PVS1 null-variant criteria under the ClinGen SVI PVS1 framework (PMC6185798).6 PS3 is not met: the only variant-specific functional study (PMID 27425891) demonstrates that hamartin(H732Y) does not produce the neuronal enlargement phenotype observed with pathogenic TSC1 stop mutants (R692X, R786X) in the same experimental system, suggesting it is not functionally equivalent to established loss-of-function variants.7 Under generic ACMG/AMP 2015 combination rules, BA1 alone meets the threshold for a Benign classification. Additionally, two strong benign criteria (BS1, BS2) independently satisfy the ≥2 Strong Benign rule for Benign.8
TSC1
Final classification
Benign
TSC1 c.2194C>T · p.His732Tyr
TSC1
BA1 is met: allele frequency of 1.38–1.40% in the European (Finnish) population in gnomAD (v2.1: 347/25116 alleles, 3 homozygotes; v4.1: 896/64030 alleles, 11 homozygotes), exceeding the 1% stand-alone benign threshold.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign; combination = 1 stand-alone benign + 2 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP4BP6
Benign
TSC1 c.2194C>T
BA1 + BS1 + BS2 + BP4 + BP6
→
Benign
4
revelbayesdelspliceai ↗
8
generic_acmg_combination_rules
Gene diagram
· NM_000368.5 · variants mapped to exon structure
TSC1
NM_000368.5
Fetching transcript structure from UCSC…
Applied criteria · 5 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
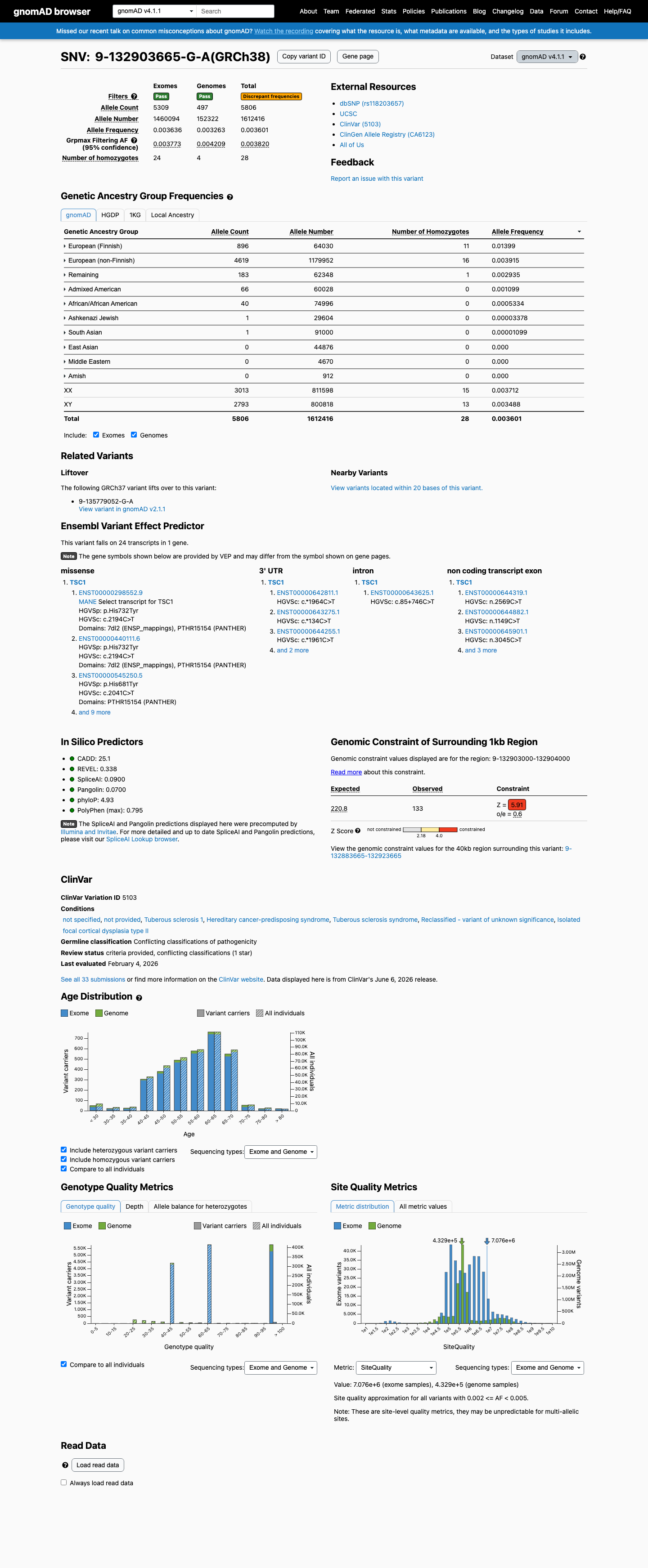
gnomAD v4.1
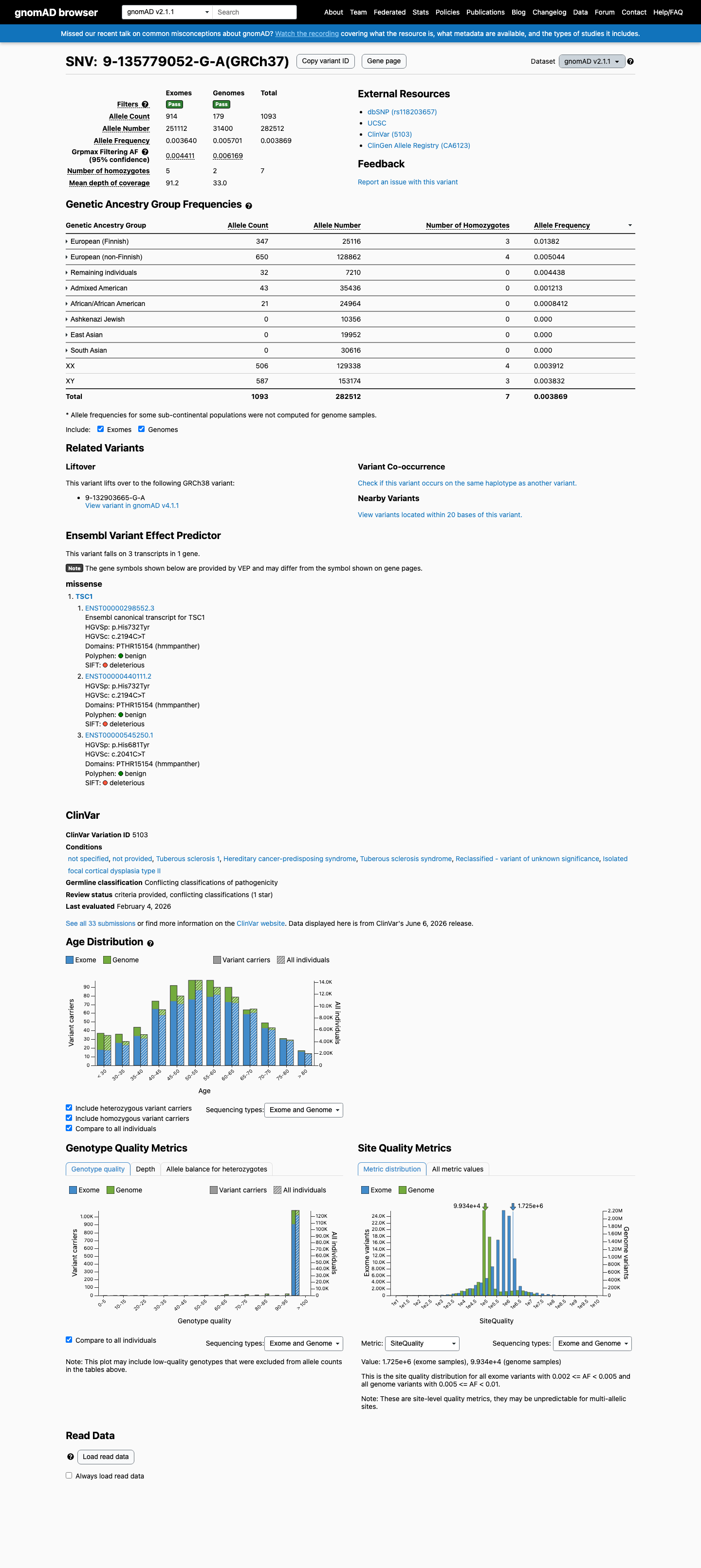
gnomAD v2.1
Population frequency
28
homozygotes observed in gnomAD v4.1. Healthy biallelic carriers are difficult to reconcile with a fully penetrant loss-of-function disease allele.
Overall AF
5806 / 1,612,416
0.36%
Highest · European (Finnish)
1.4%
Homozygotes
28
grpmax FAF
0.38%
Allele frequency by ancestry — gnomAD v4.1
observed in 6 of 9 groups
| Ancestry | Allele count | Frequency | Homozygotes |
|---|---|---|---|
| European (Finnish) | 896 / 64,030 | 1.4% | 11 |
| European (non-Finnish) | 4619 / 1,179,952 | 0.39% | 16 |
| Admixed American | 66 / 60,028 | 0.11% | 0 |
| African/African American | 40 / 74,996 | 0.053% | 0 |
| Ashkenazi Jewish | 1 / 29,604 | 0.0034% | 0 |
| South Asian | 1 / 91,000 | 0.0011% | 0 |
| Amish | 0 / 912 | — | — |
| East Asian | 0 / 44,876 | — | — |
| Middle Eastern | 0 / 4,670 | — | — |
“
This variant is present in gnomAD v4.1 (AF= 0.00360081; MAF= 0.36008%, 5806/1612416 alleles, homozygotes = 28) and has highest observed frequency in the European (Finnish) population (AF= 0.0139934; MAF= 1.39934%, 896/64030 alleles, homozygotes = 11); grpmax FAF= 0.00382016.
7
homozygotes observed in gnomAD v2.1. Healthy biallelic carriers are difficult to reconcile with a fully penetrant loss-of-function disease allele.
Overall AF
1093 / 282,512
0.39%
Highest · European (Finnish)
1.4%
Homozygotes
7
grpmax FAF
0.62%
Allele frequency by ancestry — gnomAD v2.1
observed in 5 of 8 groups
| Ancestry | Allele count | Frequency | Homozygotes |
|---|---|---|---|
| European (Finnish) | 347 / 25,116 | 1.4% | 3 |
| European (non-Finnish) | 650 / 128,862 | 0.5% | 4 |
| Remaining individuals | 32 / 7,210 | 0.44% | 0 |
| Admixed American | 43 / 35,436 | 0.12% | 0 |
| African/African American | 21 / 24,964 | 0.084% | 0 |
| Ashkenazi Jewish | 0 / 10,356 | — | — |
| East Asian | 0 / 19,952 | — | — |
| South Asian | 0 / 30,616 | — | — |
“
This variant is present in gnomAD v2.1 (AF= 0.00386886; MAF= 0.38689%, 1093/282512 alleles, homozygotes = 7) and has highest observed frequency in the European (Finnish) population (AF= 0.0138159; MAF= 1.38159%, 347/25116 alleles, homozygotes = 3); grpmax FAF= 0.00616893.
“
This variant is absent from gnomAD-Canada.

ClinVar
This variant has been reported in ClinVar as Benign (23 clinical laboratories) and as Likely benign (4 clinical laboratories). (ClinVarID = 5103)
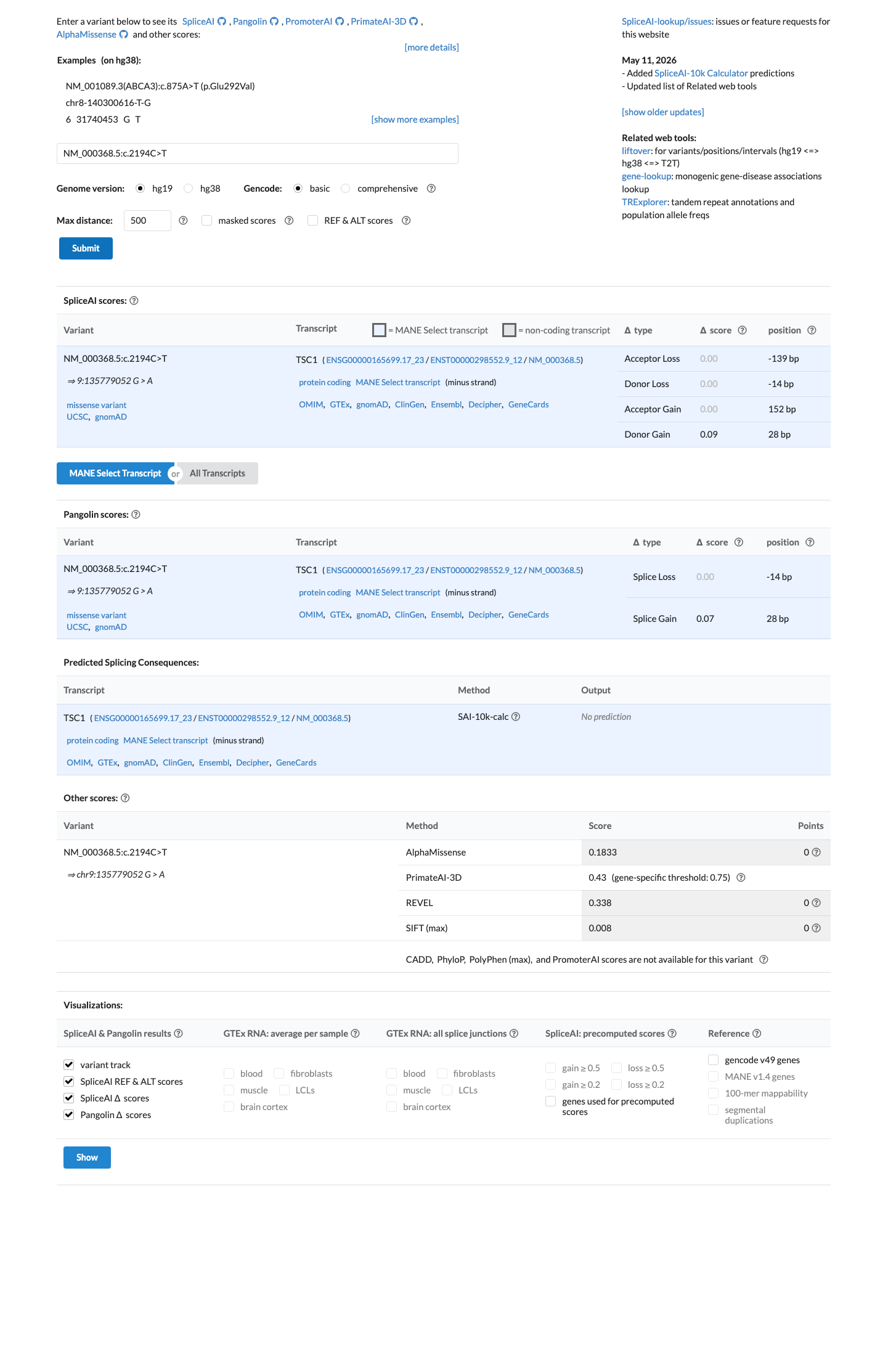
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.09). REVEL score = 0.338. BayesDel score = -0.0641334.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. TSC1, a scaffold protein, is frequently altered by mutation in bladder cancer.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV104404438, n = 6 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · 25 PMIDs triaged · 8 high-priority
25papers screened
Papers triaged by theme: functional/splicing/segregation/case_observation. high_priority_papers include abstract snippets. Use these to support PS3/BS3/PS4/PP1/PP3/PP5.
10227394 ↗
splicing rna
Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation.
Tuberous sclerosis complex is an inherited tumour suppressor syndrome, caused by a mutation in either the TSC1 or TSC2 gene. The disease is characterised by a broad phenotypic spectrum that can include seizures, mental retardation, renal dysfunction, and dermatological abnormalities. The TSC1 gene was recently identified and has 23 exons, spanning 45 kb of genomic DNA, and encoding an 8.6 kb mRNA.
BP7PP3PP5PS4PVS1
19918125 ↗
functional
Hamartin variants that are frequent in focal dysplasias and cortical tubers have reduced tuberin binding and aberrant subcellular distribution in vitro.
Focal cortical dysplasia type IIb is characterized by epilepsy-associated malformations that are often composed of balloon cells and dysplastic neurons. There are many histopathologic similarities between focal cortical dysplasia type IIb and cortical tubers in tuberous sclerosis complex (TSC), an autosomal-dominant phakomatosis caused by mutations in the TSC1 or TSC2 genes that encode hamartin an
BS3PP5PS3PS4
21309039 ↗
functional
Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex.
The effects of missense changes and small in-frame deletions and insertions on protein function are not easy to predict, and the identification of such variants in individuals at risk of a genetic disease can complicate genetic counselling. One option is to perform functional tests to assess whether the variants affect protein function. We have used this strategy to characterize variants identifie
BS3PP5PS3PS4
23514105 ↗
functional
Lack of association of rare functional variants in TSC1/TSC2 genes with autism spectrum disorder.
Autism spectrum disorder (ASD) is reported in 30 to 60% of patients with tuberous sclerosis complex (TSC) but shared genetic mechanisms that exist between TSC-associated ASD and idiopathic ASD have yet to be determined. Through the small G-protein Rheb, the TSC proteins, hamartin and tuberin, negatively regulate mammalian target of rapamycin complex 1 (mTORC1) signaling. It is well established tha
BS3PP5PS3PS4
24033266 ↗
functional
A systematic approach to assessing the clinical significance of genetic variants.
Molecular genetic testing informs diagnosis, prognosis, and risk assessment for patients and their family members. Recent advances in low-cost, high-throughput DNA sequencing and computing technologies have enabled the rapid expansion of genetic test content, resulting in dramatically increased numbers of DNA variants identified per test. To address this challenge, our laboratory has developed a s
BS3PS3PS4
25741868 ↗
functional
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
The American College of Medical Genetics and Genomics (ACMG) previously developed guidance for the interpretation of sequence variants.(1) In the past decade, sequencing technology has evolved rapidly with the advent of high-throughput next-generation sequencing. By adopting and leveraging next-generation sequencing, clinical laboratories are now performing an ever-increasing catalogue of genetic
BS3PS3PS4
26467025 ↗
functional
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
We developed a rules-based scoring system to classify DNA variants into five categories including pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign. Over 16,500 pathogenicity assessments on 11,894 variants from 338 genes were analyzed for pathogenicity based on prediction tools, population frequency, co-occurrence, segregation, and functional studies
BS3PS3PS4
27425891 ↗
functional
Minute amounts of hamartin wildtype rescue the emergence of tuber-like lesions in conditional Tsc1 ablated mice.
Tuberous sclerosis (TSC) is a phacomatosis associated with highly differentiated malformations including tubers in the brain. Those are composed of large dysplastic neurons and 'giant cells'. Cortical tubers are frequent causes of chronic seizures and resemble neuropathologically focal cortical dysplasias (FCD) type IIb. Patients with FCDIIb, however, lack additional stigmata of TSC. Mutations and
BS3PP5PS3PS4
18414213 ↗
background review
ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007.
PS3PS4
28492532 ↗
background review
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
PS3PS4
Sources & reference links